A cikk orvosi szakértője

Új kiadványok
Aper szindróma
Utolsó ellenőrzés: 04.07.2025

Minden iLive-tartalmat orvosi szempontból felülvizsgáltak vagy tényszerűen ellenőriznek, hogy a lehető legtöbb tényszerű pontosságot biztosítsák.
Szigorú beszerzési iránymutatásunk van, és csak a jó hírű média oldalakhoz, az akadémiai kutatóintézetekhez és, ha lehetséges, orvosilag felülvizsgált tanulmányokhoz kapcsolódik. Ne feledje, hogy a zárójelben ([1], [2] stb.) Szereplő számok ezekre a tanulmányokra kattintható linkek.
Ha úgy érzi, hogy a tartalom bármely pontatlan, elavult vagy más módon megkérdőjelezhető, jelölje ki, és nyomja meg a Ctrl + Enter billentyűt.

 [
[ Okoz Aper szindróma
A szindróma kialakulását a 10. kromoszómán található gén szerkezetének rendellenessége provokálja. Ez a gén felelős az ujjleválasztás folyamatáért, és ezen túlmenően a koponyavarratok időben történő lezárásáért.
Ezenkívül a terhesség alatt elszenvedett fertőző betegségek (például rubeola vagy tuberkulózis, szifilisz vagy influenza, valamint agyhártyagyulladás), valamint az anya röntgensugárzásnak való kitettsége a patológia okainak tekinthető. Leggyakrabban ilyen szindrómát észlelnek idős szülőktől született gyermekeknél.
Pathogenezis
Az Apert-szindróma örökletes. Típusa autoszomális domináns (azaz egy olyan családban, ahol a leendő szülők egyike beteg, a betegségben szenvedő baba születésének valószínűsége 50-100%).
A fibroblaszt növekedési faktor receptor 2 (FGFR2) egyedi mutációja a csontképződési útvonal mentén fejlődő progenitor sejtek számának növekedését eredményezi. Ez végső soron a szubperioszteális csontmátrix fokozott képződéséhez és a koponyavarratok korai elcsontosodásához vezet a magzati fejlődés során. A varrat olvadásának sorrendje és sebessége határozza meg a deformitás és a fogyatékosság mértékét. Miután a varratanyag meggyógyult, a varratra merőlegesen lévő egyéb szövetek növekedése korlátozott, és az összeforrt csontok egyetlen csontvázként működnek.
Az első genetikai bizonyíték arra, hogy az Apert-szindrómában a szindaktilia a keratinocita növekedési faktor receptor (KGFR) hibájának köszönhető, a fibroblasztokban a KGFR expressziója és a szindaktilia súlyossága közötti korreláció megfigyelése volt.
Az amblyopia és a kancsalság gyakoribb az FGFR2 Ser252Trp mutációval rendelkező betegeknél, míg a látóidegfő-sorvadás gyakoribb az FGFR2 Pro253Arg mutációval rendelkező betegeknél. Az FGR2 Ser252Trp mutációval rendelkező betegeknél szignifikánsan magasabb a látáskárosodás prevalenciája, mint az FGFR2 Pro253Arg mutációval rendelkező betegeknél.
Tünetek Aper szindróma
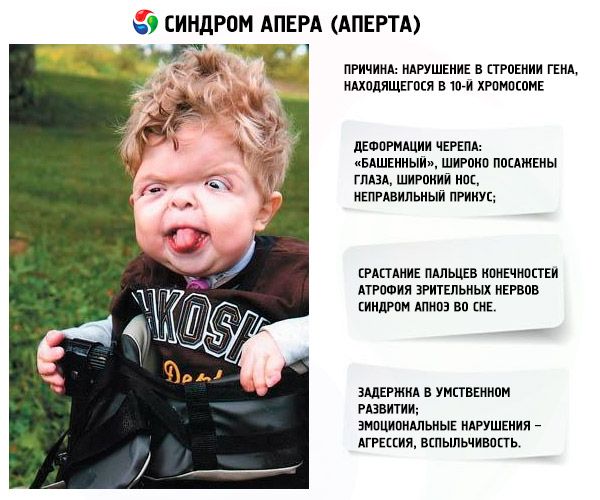
A betegség egyes megnyilvánulásai már a baba születésekor jól láthatóak, mivel az anyaméhben fejlődnek ki. A szindróma főbb tünetei közé tartoznak:
- A koponya deformálódott – magasságában megnyúlik, „torony” megjelenést kölcsönöz; emellett a szemek szélesen ülők és enyhén kidülledtek (mivel a szemüregek mérete csökken); az orr kiszélesedik és helytelen harapás alakul ki (a felső fogak túlzottan kiállnak);
- A végtagok ujjai teljesen összenőttek (főleg a gyűrűsujj, valamint a középső és a mutatóujj), és úgy néznek ki, mint egy bőrhártya vagy a csontok teljes összeolvadása; emellett extra ujjak is nőhetnek;
- Mentális retardáció (nem mindenkinél fordul elő);
- A látóidegek sorvadnak, aminek következtében csökken a látásélesség (egyes esetekben teljes látásvesztés alakul ki);
- A megnövekedett koponyaűri nyomás, amely a koponyavarratok idő előtti lezáródásának következtében jelentkezik, fejfájás és hányás formájában jelentkezik hányingerrel;
- Mivel a felső állkapocs fejletlen marad, légzési problémák merülnek fel;
- Az alvási apnoe szindróma gyakori előfordulás.
- Érzelmi megnyilvánulások – agresszió, féktelenség, erős ingerlékenység.
Komplikációk és következmények
Diagnostics Aper szindróma
A diagnózis felállításához a következő lépésekre van szükség:
- Az orvosnak elemeznie kell a beteg panaszait, valamint a kórtörténetét. Meg kell tudni, hogy voltak-e hasonló patológiás esetek a családban;
- Neurológiai vizsgálat a koponya alakjának és a beteg intellektuális fejlődésének felmérésére (speciális kérdőívek, valamint interjú);
- A szemfenék vizsgálata a megnövekedett koponyaűri nyomás tüneteinek kimutatására (a látóidegfő duzzanata, valamint széleinek elmosódása);
- A koponya állapotának felméréséhez röntgenfelvételt készítenek;
- A fej számítógépes és mágneses rezonancia képalkotása az agy és a koponya rétegenkénti szerkezetének vizsgálatára, a koponyavarratok idő előtti összenövésének tüneteinek meghatározására, valamint a hidrocephalus (a megnövekedett koponyaűri nyomás miatt felesleges agy-gerincvelői folyadék halmozódik fel (ez az agy-gerincvelői folyadék, amely elősegíti az anyagcsere-folyamatot, valamint az agy táplálkozását)) kimutatására;
- A lábak és kezek röntgenfelvétele az ujjak összenövésének okának meghatározására (ez fontos a későbbi sebészeti beavatkozás megtervezéséhez);
- Előfordulhat, hogy konzultálnak egy orvosgenetikussal és egy idegsebésszel.
Tesztek
Az FGFR2 típusú génben előforduló gyakori mutációk genetikai elemzése folyamatban van.
Megkülönböztető diagnózis
Ezt a szindrómát meg kell különböztetni más genetikai patológiáktól, amelyekben craniosynostosis figyelhető meg. Ilyen betegségek például a Pfeiffer-, Crouzon-, Saethre-Chotzen- és Carpenter-szindróma. Molekuláris genetikai vizsgálati módszereket alkalmaznak ezen rendellenességek kizárására.
Ki kapcsolódni?
Kezelés Aper szindróma
A műtétet tartják az Apert-szindróma egyetlen hatékony kezelési módjának – segít korrigálni az egyéni fizikai hibákat, és a mentális retardációt is korrigálja.
A beavatkozás során a koronális varratot zárják, hogy megakadályozzák az esetleges agysérülést. A leggyakoribb technika a craniofacialis disztrakció, amely fokozatos koponya-trakciót foglal magában. Fogszabályozási és/vagy ortognatikus műtétet végeznek az egyes archibák eltávolítására.
Ezenkívül a betegek az ujjfúzió sebészeti korrekcióján esnek át.
Referenciák
Orvosi genetika. Nemzeti Útmutató. Szerkesztette: Ginter EK, Puzyrev GEOTAR-Media alelnök. 2022