A cikk orvosi szakértője

Új kiadványok
Az osteoarthritis patogenezisének genetikai és metabolikus vonatkozásai
Utolsó ellenőrzés: 08.07.2025

Minden iLive-tartalmat orvosi szempontból felülvizsgáltak vagy tényszerűen ellenőriznek, hogy a lehető legtöbb tényszerű pontosságot biztosítsák.
Szigorú beszerzési iránymutatásunk van, és csak a jó hírű média oldalakhoz, az akadémiai kutatóintézetekhez és, ha lehetséges, orvosilag felülvizsgált tanulmányokhoz kapcsolódik. Ne feledje, hogy a zárójelben ([1], [2] stb.) Szereplő számok ezekre a tanulmányokra kattintható linkek.
Ha úgy érzi, hogy a tartalom bármely pontatlan, elavult vagy más módon megkérdőjelezhető, jelölje ki, és nyomja meg a Ctrl + Enter billentyűt.
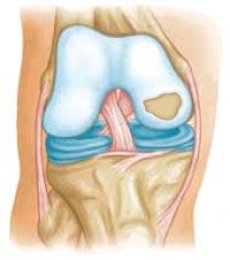
A mechanikai tényezők szerepe az osteoarthritis patogenezisében tagadhatatlan, de meggyőző bizonyítékok vannak arra, hogy az osteoarthritis egyes formái Mendel törvényei szerint öröklődnek. Az örökletes osteoartropátiák a következőkre oszthatók:
- primer generalizált osteoarthritis (PGAO),
- kristályokkal összefüggő ízületi gyulladások,
- örökletes oszteokondrodysplázia okozta korai osteoarthritis.
1803-ban W. Heberden "kissé sűrű, kis borsó méretű csomókat" írt le a kéz disztális interphalangeális ízületeinek háti felszínén. A szerző szerint ez a tünet megkülönbözteti az osteoarthritist más ízületi betegségektől, beleértve a köszvényt is. J. Hayagarth (1805) kibővítette a Heberden-csomók klinikai leírását, megjegyezve, hogy gyakran társulnak más lokalizációjú artrózissal. Később Bouchard hasonló csomókat írt le a kéz proximális interphalangeális ízületeinek háti felszínén. A "Heberden és Bouchard csomók" kifejezés használatával W. Osier megkülönböztette a "hipertrófiás ízületi gyulladást" és a "deformáló ízületi gyulladást" (1909). 1953-ban RM Stecher és H. Hersh felfedezték a Heberden-csomók előfordulását a családtagok körében, és arra a következtetésre jutottak, hogy autoszomális domináns módon öröklődnek. Az RM Stecher és H. Hersh felfedezését követő további vizsgálatok kimutatták a Heberden és Bouchard csomók összefüggését más ízületek degeneratív elváltozásaival. A klinikai vizsgálati adatok és a HLA-tipizálás alapján JS Lawrence (1977), JS Lawrence és munkatársai (1983) poligénes öröklődés jelenlétét javasolták, nem pedig egyetlen génhibát.
Az örökletes osteoarthritis fenotípusos spektruma széles skálán mozog, az enyhe formáktól, amelyek klinikailag csak késő felnőttkorban válnak nyilvánvalóvá, a nagyon súlyos, gyermekkorban manifesztálódó formákig. Hagyományosan mindezeket a formákat másodlagos osteoarthritisként osztályozták. Ma már ismert, hogy ezen fenotípusok némelyikét az ízületi porc ECM-jének makromolekuláit kódoló gének mutációi okozzák, amelyek megzavarják a porcmátrix integritását, valamint a porcsejtek proliferációjának és génexpressziójának szabályozását. Ezek az örökletes betegségek az osteoarthritis egy különálló alcsoportját képviselik, amely különbözik a másodlagos osteoarthritistől.
Az örökletes és a másodlagos osteoarthritis közötti különbségek (Williams CJ és Jimenez SA, 1999 szerint)
Örökletes osteoarthritis |
Másodlagos osteoarthritis |
|
Etiológia |
Az ízületi porcban expresszált gének mutációja |
Különböző örökletes és szerzett betegségek |
Patogenezis |
Az ízületi porc szerkezeti vagy funkcionális komponenseinek károsodása |
A betegség másodlagos megnyilvánulásai, amelyek nem mindig csak az ízületi porcot érintik |
Kezelés |
A génterápia lehetségessé teheti a génhiba korrigálását |
Az alapbetegség kezelése |
A kondrodiszplázia/oszteokondrodiszplázia klinikailag heterogén betegségek egy csoportja, melyeket az ízületi porc és a növekedési lemez növekedésének és fejlődésének rendellenességei jellemeznek. Egyes Crohn-betegségek/OCD-k osteoarthritis korai kialakulásához vezetnek, amelyet klinikailag súlyos lefolyás jellemez. Közülük a következő betegségek különböztethetők meg:
- spondyloepiphysealis diszplázia (SED),
- Stickler-szindróma,
- Knista diszplázia,
- többszörös epifízis dysplasia (MED),
- metafízis porcszöveti csomó (MCD)
- néhány oto-spondylo-meta-epiphysealis diszplázia (OSMED).
Korai kezdetű osteoarthritisben szenvedő örökletes diszpláziák (Williams CJ és Jimenez SA, 1999 szerint)
Betegség |
Lókusz |
Az öröklés típusa |
Mutált gén |
A mutáció típusa |
Korai osteoarthritis késői kezdetű SED-del (OAR)* |
12q13.1-q13.2 |
POKOL |
2. OSZLOP A, |
Bázishelyettesítés, beszúrás, deléció |
Stickler-szindróma (STL1) |
12q13.1-q13.2 |
POKOL |
COL2A1 |
Az alap cseréje, behelyezés |
Stickler-szindróma (STL2) |
6p21.3 |
POKOL |
KÓLA |
Beszúrás, törlés |
Stickler-szindróma |
1p21 |
POKOL |
KÓLA |
Az alap cseréje |
Wagner-szindróma |
12q13.1-q13.2 |
POKOL |
COUA, |
Az alap cseréje |
OSMED |
6p21.3 |
AR |
KÓLA |
Az alap cseréje |
Marshall-szindróma |
1p21 |
POKOL |
KÓLA |
Beszúrás |
Knista diszplázia |
12q13.1-q13.2 |
POKOL |
KÓLA |
Beszúrás, törlés |
M3fl(EDM1) |
19p13.1 |
POKOL |
COMP |
Az alap cseréje |
KÖZÉPES (EDM 2) |
1р32.2-рЗЗ |
POKOL |
KÓLA |
Beszúrás |
MCDS |
6q21-q22.3 |
POKOL |
KÓLA |
Bázishelyettesítés, deléció |
MCDJ Jansen |
21.2-21.3. szám |
POKOL |
PTHR, |
Az alap cseréje |
*A zárójelben a lókuszjelek szerepelnek; AD - autoszomális domináns; AR - autoszomális recesszív.
Spondyloepiphysealis diszplázia
A spondyloepiphysealis dysplasiák (SED) autoszomális domináns öröklődésű betegségek heterogén csoportját foglalják magukban, melyeket az axiális váz rendellenes fejlődése és a hosszú csőcsontok epifízisének súlyos elváltozásai jellemeznek, gyakran törpeséget okozva. Az SED-nek gyakran súlyos klinikai lefolyása van, amelyet a test és kisebb mértékben a végtagok rövidülése kísér.
Az EDS későbbi korban manifesztálódó formáiban a fenotípus gyakran alig változik, és klinikailag csak serdülőkorban jelentkezhet, amikor súlyos oszteoartrózis alakul ki. Az ágyéki gerinc deformitása a csigolyaközi porckorongok szűkületeként, platyspondyliaként és enyhe kifoszkoliozisként jelentkezhet. A perifériás ízületekben az epifízisek rendellenességei és korai degeneratív elváltozásai is észlelhetők. A perifériás ízületi károsodás legállandóbb jele a boka- és térdízületek ízületi felszíneinek ellaposodása, valamint a combcsont interkondiláris árkának ellaposodása. A combcsont fejének és nyakának rendellenességei gyakran a csípőízület oszteoartrózisának kialakulásával együtt észlelhetők, amelyek serdülőkorban jelentkeznek.
Mivel a II-es típusú kollagén a hialinporc ECM-jének fő alkotóeleme, felmerült, hogy az azt kódoló gén, a COL1A, az EDS oka. A korai osteoarthritis fenotípusa és a késői kezdetű EDS-sel összefüggő genetikai kapcsolat és a prokollagén II-es típusú gén, a COL 2 A közötti genetikai kapcsolat első leírása 1989-ből és 1990-ből származik. A késői kezdetű EDS-sel összefüggő korai osteoarthritisben szenvedő rokonoknál a COL 2 A mutációról szóló első jelentés az Arg519>Cys bázisszubsztitúciót jelentette. A mai napig további négy hasonló mutációval rendelkező családot azonosítottak. Egy másik, korai osteoarthritisben és enyhe EDS-ben szenvedő család tagjainál az Arg75>Cys bázisszubsztitúciót találták, bár az e család tagjaiban található EDS fenotípus nem hasonlít az 519-es pozícióban arginin-cisztein szubsztitúcióval rendelkező család fenotípusához. Más mutációkat, a COL 2 A-Gly976>Ser, Gly493>Ser szintén találtak az EDS-sel rendelkező családok tagjainál. J. Spranger et al. (1994) a "11-es típusú kollagénopátia" kifejezést a porcszövet örökletes betegségeinek leírására használta, amelyek a COL1A prokollagén II-es típusú génjének primer mutációjával járnak.
A Stickler-szindróma klasszikus formája
A kórképet először 1965-ben írta le GB Stickler és kollégái, akik örökletes arthro-ophthalmopathiának nevezték el. A GB Stickler által leírt szindrómát látáskárosodás és súlyos degeneratív ízületi betegség jellemezte, amely általában az élet harmadik vagy negyedik évtizedében alakul ki. Autoszomális domináns módon öröklődő rendellenesség, amelynek előfordulási gyakorisága körülbelül 10 000 élveszületésből 1. A klinikai tünetek közé tartozik a rövidlátás, a progresszív süketség, a szájpadhasadék, az állkapocs hypoplasiája (Pierre-Robin anomália) és az epiphysis hypoplasiája. Az újszülöttkori időszakban a Stickler-szindrómás betegek röntgenfelvételei megnagyobbodott epiphysiseket mutatnak, elsősorban a proximális combcsontot és a disztális sípcsontot. A növekedés során epiphysis dysplasia alakul ki, amely az epiphysisek szabálytalan elcsontosodásában és az azt követő degeneratív változásokban nyilvánul meg.
Mivel a COL 2 A az ízületi porcban és a szemgolyó üvegtestében expresszálódik, a Stickler-szindróma előfordulása összefüggésbe hozható volt a gén patológiájával. Több Stickler-szindrómás család vizsgálata azonban kimutatta, hogy nem minden családban fordul elő a COL 2 A-val összefüggő betegség. A betegségnek ezt a formáját I. típusú Stickler-szindrómának nevezik (lókuszjel STL1).
A Stickler-szindróma klinikai manifesztációinak spektruma széles skálán mozog, és a mai napig számos fenotípust azonosítottak. Ezek közé tartozik a Wagner-szindróma, amelyet a szemgolyó károsodásának túlnyomó többsége jellemez; a Wagner-szindrómában az oszteoartritisz gyakorlatilag soha nem alakul ki, bár a COL2A gén mutációját ( Gly67>Asp bázisszubsztitúció) azonosították a betegeknél. Továbbra sem világos, hogy egy ilyen COL-mutáció miért csak az üvegtest működését károsítja, és miért nem érinti a hialinporcot.
A Stickler-szindróma egy másik formája az úgynevezett holland variáns; a látáskárosodás kivételével a szindróma összes klasszikus megnyilvánulása jellemzi. HG Brunner és munkatársai (1994) kimutatták, hogy a Stickler-szindróma holland fenotípusa a COL,,A2 génben lévő mutációval társul: a domináns mutáció egy 54 bázispár hosszúságú deléció, amelyet egy exon deléció követ. M. Sirko-Osadsa és munkatársai (1998) egy másik, a korábbi szerzők által leírttól független családról számoltak be, amely hasonló fenotípussal és a COL,,A2 génben lévő mutációval (27 bázispár hosszúságú deléció) rendelkezik , ami megerősíti HG Brunner és munkatársai (1994) adatait. Ezt a variánst II. típusú Stickler-szindrómának nevezik (lókuszszimbólum STL1).
Nemrégiben a Stickler-szindróma egy harmadik lókuszát azonosították egy olyan család tagjaiban, ahol üvegtesti és retina patológia mutatkozott, és ezek a változások fenotípusosan szignifikánsan eltérnek a szindróma "klasszikus" változatában megfigyelt változásoktól. A család tagjainál a COL2A| gén mutációját (Gly97>Val bázisszubsztitúció) találták. Természetesen a Stickler-szindróma ezen feno- és genotípusú eseteinek új leírására van szükség AJ Richards és munkatársai eredményeinek megerősítéséhez.
A Marshall-szindróma és a Stickler-szindróma klasszikus változata közötti nozológiai összefüggést már régóta vitatják. A Marshall-szindrómát ma különálló fenotípusként osztályozzák, főként az arckoponya kifejezettebb deformációja miatt, bár a perifériás ízületek károsodása hasonló az I. típusú Stickler-szindrómához. Marshall-szindrómában a térdízületek és a lumbosacralis gerinc osteoarthritisa 30 év után kezdődik. A szindróma oka a IX-es típusú kollagén gén, a COL n A1 mutációja.
 [ 1 ], [ 2 ], [ 3 ], [ 4 ], [ 5 ], [ 6 ]
[ 1 ], [ 2 ], [ 3 ], [ 4 ], [ 5 ], [ 6 ]
OSMED
Ezt a fenotípust egy holland családban írták le, ahol serdülőkorban degeneratív elváltozások jelentkeztek az ízületekben, amelyek osteoarthrosisra hasonlítottak, és főként a csípő-, térd-, könyök- és vállízületeket érintették; sajátos arcvonásokat, fokozott ágyéki lordózist, megnagyobbodott interphalangeális ízületeket és halláskárosodást is észleltek, de látási rendellenességeket nem észleltek (Vikkula M. et al., 1995). A kutatók mutációt találtak a II. típusú kollagén, a COL,,A 2 α2 -láncát kódoló génben.
Knista diszplázia
Jellemzői a törzs és a végtagok rövidülése, az arc és az orrnyereg ellaposodása, exophthalmus és súlyos ízületi rendellenességek. A Kniest-szindrómában szenvedő betegeknél az ízületek, amelyek általában születésüktől fogva nagyok, gyermekkorban és kora serdülőkorban is tovább növekednek. Gyakran előfordul náluk rövidlátás, halláskárosodás, szájpadhasadék és dongaláb is; a legtöbb betegnél korán súlyos degeneratív elváltozások alakulnak ki, különösen a térd- és csípőízületekben kifejezve. A gerincröntgenfelvételek a csigolyatestek és a platyspondylia ellaposodását és jelentős megnyúlását mutatják. A hosszú csőcsontok súlyzószerűen deformáltak, az epiphysisek csontosodása lassú. A kéz ízületeiben az epiphysisek ellaposodtak, az ízületi rések pedig szűkültek. Az ízületi porc puha, rugalmassága csökkent; szövettanilag nagy ciszták találhatók benne (a "svájci sajt" tünete). A Kniest-szindrómát a prokollagén II. típusú COb2A1 génjének mutációja okozza.
[ 7 ], [ 8 ], [ 9 ], [ 10 ], [ 11 ], [ 12 ], [ 13 ], [ 14 ]
Többszörös epifízis dysplasia (MED)
Heterogének betegségcsoport, melyet a hosszú csőcsontok növekedési lemezeinek rendellenes fejlődése, valamint a korai (gyermekkorban jelentkező) súlyos oszteoartrózis jellemez, amely mind az axiális, mind a perifériás ízületeket (leggyakrabban a térd-, csípő-, váll- és kézízületeket) érinti. Klinikailag a MED ízületi fájdalomként és merevségként, valamint a járás megváltozásában nyilvánul meg. A MED-ben szenvedő betegeknél minimális változások figyelhetők meg a gerincoszlopban (a csigolyatestek különböző mértékű ellaposodása), néha a gerinc ép marad. A betegek alacsony termete is jellemző, bár a törpeség ritkán alakul ki. A látószervet nem érinti. A MED számos változatot foglal magában, például a Fairbanks és Ribbing fenotípust.
A MED-ek autoszomális domináns módon öröklődnek, változó penetranciával. Mivel a MED-ek jellemzője az epifízis növekedési lemez rendellenessége, felmerült, hogy ezeket a diszpláziákat a növekedési lemez porcának makromolekuláit kódoló gének hibája okozza. Kiderült, hogy legalább három lókusz kapcsolódik a MED fenotípushoz. EJ Weaver és munkatársai (1993), JT Hecht és munkatársai (1992) tanulmányai kizárták a II. és VI. típusú kollagén génjeit, a proteoglikánok magfehérjéjét és a porc kötőfehérjéjét a MED-ek "bűnöseinek" listájáról. JT Hecht és munkatársai (1993), R. Oehelmann és munkatársai (1994) összefüggést találtak a MED, valamint a klinikailag összefüggő pszeudoachondroplasia szindróma és a 19-es kromoszóma pericentromer régiója között. Későbbi vizsgálatok mutációt azonosítottak a porc oligomer mátrixfehérjét (OMMP) kódoló génben három MED-es betegnél (lókusz szimbólum EDM1). Mivel mindhárom mutáció az OMMP kalciumkötő doménjét kódoló gén régiójában történt, valószínű, hogy e fehérje kalciumkötő funkciója elengedhetetlen a növekedési lemez porcának normális fejlődéséhez.
MD Briggs és munkatársai (1994) egy holland családról számoltak be, ahol a MED fenotípus az 1. kromoszóma egy olyan régiójához kapcsolódik, amely a IX. típusú kollagéngén egyikét, a COL1A1-et (az EDM 2 lókusz szimbóluma) tartalmazza. Figyelemre méltó, hogy a talált mutáció volt az első bizonyíték arra, hogy a IX. típusú kollagén, amely a II. kollagén fibrillumok felszínén lokalizálódik, szerepet játszik a hialinporc integritásának fenntartásában. M. Deere és munkatársai (1995) kimutatták, hogy a Fairbanks fenotípus genetikailag nem társult sem az EDM, sem az EDM2 lókuszhoz, ami megerősíti a MED heterogenitását.
Metafízis porcszöveti kondrodiszplázia (MCD)
A hialinporc örökletes betegségeinek heterogén (több mint 150 típusát írták le) csoportja, amelyek klinikailag korai oszteoarthrózisként jelentkeznek. Az MHD-ket a csontmetafízis változásai jellemzik. Klinikailag alacsony termet, megrövidült végtagok, görbe sípcsont és „kacsa” járás formájában jelentkeznek. Az MHD-ben szenvedő betegek más rendszerek (például az immun- és az emésztőrendszer) károsodásának jeleit is mutatják. A növekedési lemez porcának dezorganizációja figyelhető meg, ami szövettanilag proliferált és hipertrófiás porcsejtek csoportjaiban nyilvánul meg, amelyeket megvastagodott szeptumok és dezorganizált mátrix vesz körül, valamint a nem meszesedett porc behatolása a szubkondrális csontba.
A Jansen-, Schmid- és McKusick-szindrómák a legtöbbet tanulmányozott MHD-k. A csontvázrendszeri rendellenességek jellemzőiben hasonlóak, de súlyosságukban különböznek (Jansen-szindróma-McKusick-szindróma-Schmid-szindróma). A leggyakoribb a Schmid-szindróma (az MCDS lókusz szimbóluma), amely autoszomális domináns módon öröklődik. Radiológiailag a szindróma coxa vara, a csőcsontok rövidülése és görbülete, a metafízisek csésze alakú deformációja (a combcsont proximális, mint disztális részén kifejezettebb) formájában jelentkezik. A legkifejezettebb változások a hosszú csőcsontok növekedési lemezein figyelhetők meg.
Legalább 17 különböző típusú kollagén X génmutációt írtak le Schmid-szindrómás betegeknél. A kollagén X a növekedési lemezek hipertrófiás porcsejtjeiben expresszálódik, és szerepet játszhat a csontosodási folyamatokban. Így a COb2A1 kollagén X gén mutációja a Schmid-szindróma legvalószínűbb oka.
A Jansen-szindrómás gyermekeknél hiperkalcémia, emelkedett vizelet-foszfátszint, valamint csökkent mellékpajzsmirigy-hormon (PTH) és PT-vel kapcsolatos peptidszint figyelhető meg. Ez utóbbi anomáliája valószínűleg felelős a Jansen-szindróma kialakulásáért. 1994-ben AS Karaplis és munkatársai publikálták egy eredeti tanulmány eredményeit. Miután egérembrionális őssejtekben a PT-vel kapcsolatos peptidet kódoló gént károsították, az ebben az allélban hiányos egerek születésük után azonnal elpusztultak. A szubkondrális csontfejlődés rendellenességét, a porc növekedésének zavarát és a kondrocita-proliferáció csökkenését találták náluk. 1995-ben E. Schipani és munkatársai heterozigóta mutációról számoltak be a PTH receptor génjében egy Jansen-szindrómás betegnél. A mutáció egy Gys223>Arg bázisszubsztitúcióból állt, ami a cAMP felhalmozódásához vezetett; Ez azt jelenti, hogy a 223-as pozícióban lévő hisztidin aminosav kulcsszerepet játszik a jelátvitelben. Később E. Schipani és munkatársai... (1996) további három Jansen-szindrómás betegről számolt be, akik közül kettőnek hasonló mutációja volt, a harmadiknak pedig TrА10>Ро szubsztitúciója.
Elsődleges generalizált osteoarthritis
Az osteoarthritis leggyakoribb örökletes formája a primer generalizált osteoarthritis (PGOA), amelyet először JH Kellgren és R. Moore írt le különálló nozológiaként 1952-ben. Klinikailag a primer generalizált osteoarthritist a Bouchard és Heberden csomók, a poliartikuláris elváltozások megjelenése jellemzi. A primer generalizált osteoarthritist az osteoarthritis manifesztációjának korai kezdete és gyors progressziója jellemzi. Radiológiailag a primer generalizált osteoarthritis nem különbözik a nem örökletes osteoarthritistől. Annak ellenére, hogy a primer generalizált osteoarthritis etiopatogenezisének kérdése még mindig vitatott, a vizsgálatok bizonyítják az örökletes hajlam fontos szerepét a primer generalizált osteoarthritis kialakulásában és progressziójában.
Így JH Kellgren és munkatársai (1963) a férfi rokonok 36%-ánál és a női rokonok 49%-ánál találtak Boucharay-Heberden csomókat, míg az átlagpopulációban ezek az arányok 17, illetve 26% voltak. Primer generalizált osteoarthritisben szenvedő egyéneknél gyakrabban észlelhető a HLA A1B8 haplotípus és az a1-antitripszin MZ izoformája. Egy klasszikus, ikreket vizsgáló vizsgálatban TD Spector és munkatársai (1996) 130 monozigóta és 120 kétpetéjű női ikerpár térd- és kézízületeinek röntgenvizsgálatát végezték az osteoarthritisre jellemző elváltozások kimutatására. Kiderült, hogy az osteoarthritis radiográfiai jeleinek egyezése az összes lokalizáció esetében kétszer magasabb volt a monozigóta ikrekben a kétpetéjű ikrekhez képest, és a genetikai tényezők hozzájárulása 40-70% között mozgott. GD Wright és munkatársai noduláris osteoarthritisről szóló tanulmánya... (1997) kimutatta a betegség korai kezdetét, magas súlyosságát, valamint negatív korrelációt a betegeknél a betegség kezdetének életkora és a szüleik fogantatásának életkora között.
A kristályokkal összefüggő ízületi betegségek közül a húgysavkristályok és a kalciumtartalmú kristályok ízületi üregben való lerakódásának családi hajlama van.
Örökletes kristályhoz társuló ízületi gyulladások (Williams CJ és Jimenez SA, 1999 szerint)
Betegség |
Lókusz |
Az öröklés típusa |
Mutált gén |
A mutáció típusa |
Köszvény (HPRT)* |
Xq27 |
X-hez kötött |
HPRT1 |
Bázishelyettesítés, deléció |
Köszvény (PRPS) |
Xq22-q24 |
X-hez kötött |
PRPS1 |
Az alap cseréje |
Elsődleges pirofoszfát ízületi gyulladás (CCAL1) |
5p15.1-p15.2 |
POKOL |
? |
? |
Korai kezdetű pirofoszfát ízületi gyulladás, amely 0A-val (CCAL2) társul |
8q |
POKOL |
? |
? |
*A zárójelben a lókuszjelek szerepelnek; AD – autoszomális domináns.
1958-ban D. Zintann S. Sitaj klinikai leírásokat közölt egy általuk „kondrokalcinosisnak” nevezett patológiáról 27 beteg esetében. A betegek többsége öt családhoz tartozott, ami örökletes komponensre utal a betegség etiopatogenezisében. Később D. McCarty és J. L. Hollander (1961) két olyan betegről számoltak be, akiknél köszvény gyanúja merült fel, és nem urát kristályok rakódtak le az ízületi üregben. A röntgenvizsgálat számos ízület hialinporcának rendellenes meszesedését mutatta ki.
Radiográfiailag a kalcium-pirofoszfát-dihidrát kristálylerakódási betegség, vagy pirofoszfát artropátia, hasonlít a sporadikus oszteoartritiszre, de gyakrabban érinti azokat az ízületeket, amelyek nem jellemzőek az oszteoartrózis gyakori formáira (pl. metakarpofalangeális, scaphoradiális, patellofemorális térdízületek). Pirofoszfát artropátia esetén gyakrabban képződnek szubkondrális csontciszták. Bár a legtöbb esetben a kondrokalcinozis a másodlagos oszteoartrózis manifesztációja előtt jelentkezik, egyes egyéneknél a betegség idiopátiás oszteoartrózisként kezdődhet, amelyet anyagcserezavarok (hemokromatózis, hiperparatireózis, hipomagnesémia stb.) kísérnek.
Valószínűleg az ízületi porc ECM-jében bekövetkező szerkezeti változások idézik elő a kalcium-pirofoszfát-dihidrát kristályok lerakódását. AO Bjelle (1972, 1981) a kollagéntartalom csökkenését és a kollagénrostok fragmentációját találta a pirofoszfát artropátiában szenvedő svéd családtagok ízületi porc mátrixának középső zónájában. Mivel ezek a területek nem tartalmaztak kristályokat, a szerzők azt feltételezték, hogy a leírt mátrix-anomália hajlamosíthat a lerakódásukra és az ízületekben bekövetkező degeneratív elváltozások kialakulására. A pirofoszfát artropátia szórványos eseteinek tanulmányozása alapján K. Ishikawa és munkatársai (1989), I. Masuda és munkatársai (1991) arra a következtetésre jutottak, hogy a chondrocalcinosist az ECM fehérjéket kódoló gének mutációja okozza. CJWilliams és munkatársai (1993), AJ Reginato és munkatársai... (1994) egy nagy család tagjainál egy heterozigóta COL2A mutációt (bázisszubsztitúció Argl5>Cys) talált, ahol a klinikai fenotípus súlyos korai osteoarthritis volt ankylosissal, késői spondyloepiphysealis dysplasia kialakulásával és a hialin- és fibroporc chondrocalcinosisával. Kiderült azonban, hogy e család tagjainál a chondrocalcinosis az OA másodlagos oka volt.
Azt is felvetették, hogy az ECM szervetlen komponensei hozzájárulnak a kristályképződéshez. Például a hipomagnesémia chondrocalcinosist okoz a pirofoszfatáz enzim gátlásával, ami viszont csökkenti a kristályok oldódását. A pirofoszfát arthropathiában szenvedő betegek ízületi folyadékában emelkedett szervetlen foszfátszintet találtak. Ez és más megfigyelések arra utalnak, hogy a pirofoszfát arthropathiában szenvedő betegeknél lokális pirofoszfát-anyagcserezavar áll fenn. Leírták a nukleozid-trifoszfát pirofoszfohidroláz enzimet, amely szerepet játszhat a pirofoszfát kristályok képződésében az ECM-ben való lerakódásuk területén. Ennek az enzimnek a szintjének emelkedését találták szórványos pirofoszfát arthropathia esetekben, de ezt a rendellenességet nem figyelték meg a betegség familiáris formáiban (Ryan LM et al., 1986). Azonban a familiáris pirofoszfát arthropathiában szenvedő betegek fibroblasztjainak és limfoblasztjainak tenyésztésekor a szervetlen foszfátok tartalmának növekedését észlelték, ami szintén megerősíti azt a feltételezést, hogy a lokális pirofoszfát-anyagcsere zavarai szerepet játszanak a betegség patogenezisében.
Az utóbbi években kísérleteket tettek a pirofoszfát artropátia családi előfordulásáért „bűnös” gének azonosítására. Így egy nagycsalád (Maine, USA) tagjaitól származó genetikai anyag elemzése, amelyben a chondrocalcinosis súlyos, gyorsan progrediáló, nem diszpláziás osteoarthrosis következtében alakult ki, kizárta a betegség és a COL 2 lókusz közötti kapcsolatot. A jelen tanulmány szerzői azonban összefüggést találtak a vizsgált pirofoszfát artropátia fenotípusa és a 8-as kromoszóma hosszú karján található lókusz (a CCAL lókusz szimbóluma) között. AG Hughes és munkatársai (1995) összefüggést találtak egy brit család primer chondrocalcinosis fenotípusa és a CCAL1 lókusz között, amely az 5-ös kromoszóma rövid karján, az 5p15 régióban lokalizálódik. CJ Williams és munkatársai (1996) szerint egy argentin család tagjainál a CCAL1 lókusz valamivel proximálisabban helyezkedett el, mint az előző esetben, az 5p15.1 régióban. Hasonló genotípust találtak egy franciaországi család tagjainál.
Így a leírt vizsgálatok adatai arra utalnak, hogy a pirofoszfát artropathia familiáris formája klinikailag és genetikailag heterogén betegség, amelyet legalább három különböző gén mutációi okozhatnak.