A cikk orvosi szakértője

Új kiadványok
Xeroderma pigmentosum
Utolsó ellenőrzés: 04.07.2025

Minden iLive-tartalmat orvosi szempontból felülvizsgáltak vagy tényszerűen ellenőriznek, hogy a lehető legtöbb tényszerű pontosságot biztosítsák.
Szigorú beszerzési iránymutatásunk van, és csak a jó hírű média oldalakhoz, az akadémiai kutatóintézetekhez és, ha lehetséges, orvosilag felülvizsgált tanulmányokhoz kapcsolódik. Ne feledje, hogy a zárójelben ([1], [2] stb.) Szereplő számok ezekre a tanulmányokra kattintható linkek.
Ha úgy érzi, hogy a tartalom bármely pontatlan, elavult vagy más módon megkérdőjelezhető, jelölje ki, és nyomja meg a Ctrl + Enter billentyűt.
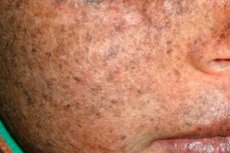
A Xeroderma pigmentosum egy autoszomális recesszív öröklődésű DNS-javító genetikai rendellenesség. A betegséget az ultraibolya sugárzás és más mérgező anyagok által károsított DNS javításában részt vevő gének mutációi okozzák.
Leggyakrabban ez a betegség gyermekeket érint, akiket "az éjszaka gyermekeinek" is neveznek. A betegség gyakori szövődményei a bazálissejtes karcinóma és más rosszindulatú bőrdaganatok, az áttétes rosszindulatú melanoma és a laphámsejtes karcinóma.
 [ 1 ]
[ 1 ]
Pathogenezis
A betegség kialakulásában fontos szerepet játszik az UV-endonukleáz enzimek hiánya a beteg sejtjeiben (fibroblasztokban), vagy teljes hiányuk. Ez az enzim felelős az ultraibolya sugarak által károsított DNS reprodukciójáért. Más források szerint a DNS-polimeráz-1 enzim hiánya játszik fontos szerepet a betegség kialakulásában egyes betegeknél. A betegség leggyakrabban 280-310 nm hullámhosszú sugarak hatására alakul ki. Úgy vélik, hogy a pigment xeroderma különféle fotodinamikus anyagok szervezetbe jutása vagy az emberi biológiai környezetben megnövekedett porfirinszint miatt alakul ki.
A betegség korai szakaszában hiperkeratózis, az epidermisz fókuszának sorvadása, a Malpighi-réteg elvékonyodása, a melanin granulátumok számának növekedése a sejtek bazális rétegében, valamint krónikus gyulladásos infiltráció figyelhető meg a dermis felső rétegében (főleg az erek körül). Ezt követően degeneratív változásokat észlelnek a kollagén- és elasztikus rostokban, és a tumor stádiumában a bőrrákra jellemző jelek mutatkoznak.
Tünetek xeroderma pigmentosum
A betegség férfiakat és nőket egyaránt érint. A betegség kora gyermekkorban, tavasszal vagy nyáron kezdődik. A beteg bőre különösen érzékeny a napfényre. Ezért az első kiütések hiperpigmentáció formájában, anyajegyekhez hasonlóan, a napfénynek kitett bőrfelületeken jelennek meg. A kiütés fokozódik, az eritéma fokozódik, sötétbarnává válik, teleangiektázia, angioma és atrófia jelenik meg. Ezt követően papillomák és szemölcsök nőnek (főleg az arc és a nyak bőrén), foltok és fekélyek jelennek meg. A fekélyes hegesedés következtében az orr elvékonyodik ("madárcsőr"), a szemhéj kifelé fordul. A papillomák általában rosszindulatú daganatokká fejlődnek. A pigmentált xeroderma általában spinocelluláris rákkal, melanoszarkómákkal együtt fordul elő.
Mi bánt?
Szakaszai
A xeroderma pigmentosum klinikai lefolyásában öt stádiumot különböztetünk meg: gyulladásos (erythemás), hiperpigmentáció, atrófia, hiperkeratózis és rosszindulatú daganatok.
A gyulladásos (erythemás) stádiumban duzzanat, vörös foltok, néha hólyagok és hólyagok jelennek meg a napfénynek kitett bőrfelületeken (arc, nyak, mellkas felső része, karok, kezek). A napfénynek nem kitett területeken szinte egyáltalán nincs kiütés.
Hiperpigmentáció esetén a vörös foltok helyén világosbarna, barna, anyajegyekhez hasonló hiperpigmentált foltok jelennek meg.
Sorvadás esetén a bőr kiszárad, elvékonyodik és ráncosodik. Számos apró, csillag alakú telangiektázia és fényes felszínű heg figyelhető meg az ajkak és az orr bőrén. A sorvadás és a hegek miatt mikrotómia (a szájnyílás csökkenése), ektropium, a fülek és az orr elvékonyodása, az orrnyílás és a száj atresiája alakul ki. A betegek 80-85%-ánál a szemek is érintettek: kötőhártya-gyulladás, keratitisz, a szaruhártya és a nyálkahártya károsodása, valamint látásgyengeség figyelhető meg. A szemhéjak bőrén diszkrómia, telangiektázia, hiperkeratózisos elváltozások és daganatok figyelhetők meg.
Hiperkeratózis esetén a fent leírt kóros gócokban szemölcsös daganatok, papilloma, keratoakantoma, fibroma, angiofibrioma és más jóindulatú daganatok jelennek meg. Ezért egyes tudósok a pigment xerodermát a rákmegelőző betegségek csoportjába sorolják.
A betegség kezdetétől számított 10-15 év elteltével rosszindulatú bőrdaganatok (basalioma, endothelioma, angiosarcoma) jelennek meg atrófiásan megváltozott gócokban és pigmentfoltokban. A daganatok rövid időn belül elpusztulnak, áttétet képeznek a belső szervekben, és halálhoz vezetnek. Egyes betegek belső szerveiben és szöveteiben általános disztrófiás elváltozások figyelhetők meg (a második és harmadik lábujj szindaktéliája, fogdisztrófia, teljes hajhullás stb.).
Forms
A xeroderma pigmentosum neurológiai formája két szindrómában nyilvánul meg.
A Reed-szindrómát a xeroderma pigmentosum és a mikrokefália klinikai tünetei, az idiopathia és a csontvázrendszer lassú növekedése jellemzi. A betegség neurológiai formájában a DNS-reparáció nehézkes, és a röntgenterápia a fő kóros gócok növekedéséhez vezet.
A De Sinctis-X Cocchione szindrómát a következő klinikai tünetek jellemzik:
- a xeroderma pigmentosum klinikai tüneteinek kialakulása különösen fényérzékeny bőrön;
- rosszindulatú daganatok korai megjelenése;
- spasztikus bénulás, mikrocefália és demencia egyidejű lefolyása;
- veleszületett deformitások és törpeség;
- a gonádok fejletlensége;
- gyakori vetélések;
- recesszív átvitel öröklés útján.
Egyes bőrgyógyászok szerint a Santis-Cacchione szindróma nem önálló betegség, hanem a xeroderma pigmentosum súlyos és teljes mértékben kifejezett klinikai megnyilvánulása.
Genetikai formák
Típusok |
Gén |
Lókusz |
Leírás |
A, I, XPA típus |
XPA |
9q22.3 |
Xeroderma pigmentosum (XP) A csoport – klasszikus forma |
B, II, XPB típus |
XPB |
2q21 |
XP B csoport |
C típus, III, XPC |
XPC |
3p25 |
XP C csoport |
D, IV, XPD típus |
XPD ERCC6 |
19q13.2-q13.3, 10q11 |
XP D csoport vagy De Sanctis-Cacchione szindróma |
E, V, XPE típus |
DDB2 |
11p12-p11 |
XP E csoport |
F, VI, XPF típus |
ERCC4 |
16p13.3-p13.13 |
XP F csoport |
G, VII, XPG típus |
RAD2ERCC5 |
13q33 |
XP G csoport és COFS szindróma (cerebro-oculo-facioskeletális szindróma) 3-as típus |
V. típus, XPV |
POLH |
6p21.1-p12 |
Xeroderma Pigmentosa variáns |
Mit kell vizsgálni?
Hogyan kell megvizsgálni?
Ki kapcsolódni?
Kezelés xeroderma pigmentosum
A maláriaellenes gyógyszerek (delagil, plakenil, resokvin stb.), amelyek védik a DNS-t az ultraibolya sugaraktól, gátolják a depolimerizációs folyamatot, csökkentik a bőr érzékenységét (fotoszenzibilizációját) a napfényre. Ezek a gyógyszerek gyulladáscsökkentő és hiposzenzibilizáló tulajdonságokkal rendelkeznek. Általános terápiát ajánlott vitaminterápiával (B1, B2, PP, B6, B12, A, E), antihisztaminokkal (tavegil, difenhidramin, szuprastin), deszenzibilizáló szerekkel (nátrium-tioszulfát, 10% kalcium-klorid intravénásan 10 ml) együtt végezni.
Helyi kezelésre fényvédő krémeket és kenőcsöket használnak.
A pigment xeroderma tumoros formájában sebészeti módszereket, folyékony nitrogént és lézersugarakat alkalmaznak. A napfénytől való védelem érdekében laza ruházatot, napvédő sapkát és kesztyűt kell viselni.
Előrejelzés
A betegek többsége (2/3) 15 éves kora előtt meghal. Egyes betegek 40-50 évig is élhetnek.
[ 20 ]