A cikk orvosi szakértője

Új kiadványok
Angelman-szindróma gyermekeknél és felnőtteknél
Utolsó ellenőrzés: 04.07.2025

Minden iLive-tartalmat orvosi szempontból felülvizsgáltak vagy tényszerűen ellenőriznek, hogy a lehető legtöbb tényszerű pontosságot biztosítsák.
Szigorú beszerzési iránymutatásunk van, és csak a jó hírű média oldalakhoz, az akadémiai kutatóintézetekhez és, ha lehetséges, orvosilag felülvizsgált tanulmányokhoz kapcsolódik. Ne feledje, hogy a zárójelben ([1], [2] stb.) Szereplő számok ezekre a tanulmányokra kattintható linkek.
Ha úgy érzi, hogy a tartalom bármely pontatlan, elavult vagy más módon megkérdőjelezhető, jelölje ki, és nyomja meg a Ctrl + Enter billentyűt.

Számos olyan betegség van, amelynél az olyan kifejezések, mint a „vigyázz magadra, és nem leszel beteg”, legalábbis nevetségesen hangzanak. Ezek olyan kórképek, amelyekben a gyermek testében már a születés előtt is rejlenek bizonyos mentális és fizikai rendellenességek, de a szülők nem hibáztathatók ezért. Az ilyen betegségeket a kromoszómakészletek mutációi vagy rendellenességei okozzák, és kromoszómális vagy genetikai betegségeknek nevezzük őket. Angelman-szindróma, Down-szindróma, Patau-szindróma, Edwards-szindróma, Turner-szindróma, Prader-Willi-szindróma - ez csak egy része a genetikai betegségeknek egy meglehetősen tisztességes listából.
Boldog ember szindróma
Ezúttal az angol gyermekorvosról, Harry Angelmanról elnevezett patológiáról fogunk beszélni, aki 1965-ben először vetette fel a probléma kérdését, miután előző nap három szokatlan gyermekkel találkozott a praxisában, akiket közös, sajátos tünetek egyesítettek. Az orvos babagyerekeknek nevezte el ezeket a gyerekeket, és írt róluk egy cikket, amelyet kezdetben „Bábgyerekek” címmel írtak. Maga a cikk és a címe egy veronai múzeumokban látott festmény hatására íródott. A festmény egy nevető fiút ábrázolt, és „A bábfiú” címet kapta. A festményen ábrázolt gyermek és a három gyermek közötti kapcsolat, akikkel Angelman egykor a praxisában találkozott, arra késztette a gyermekorvost, hogy a gyerekeket egy csoportba vonja össze a betegségük miatt.
Nincs abban semmi meglepő, hogy a cikkben említett gyerekeket más orvosok nem vették észre. Végül is első pillantásra úgy tűnt, hogy teljesen más betegségeik vannak, annyira eltérő volt a betegség általános klinikai képe 3 különböző esetben. Talán az "új" kromoszóma-patológia más tudósokat is érdekelt volna, de akkoriban a genetika még nem volt annyira fejlett, hogy megerősítse az angol orvos hipotézisét. Ezért, miután bizonyos érdeklődés mutatkozott iránta, a cikk hosszú időre a hátsó polcra került.
Az Angelman-szindróma következő említése, amelyre az angol gyermekorvos, G. Angelman cikke ekkoriban hivatkozott, a 20. század 80-as éveinek elejére nyúlik vissza. És csak 1987-ben sikerült megtalálni az okát annak, hogy a gyermekek egy kis része miért születik olyan eltérésekkel, hogy kívülről úgy tűnik, folyamatosan mosolyognak és boldogok. Valójában ez egyáltalán nem igaz, és a mosoly csak egy grimasz, amely mögött egy boldogtalan emberi lélek és a szülők fájdalma rejtőzik.
Járványtan
A statisztikák szerint a gyermek kromoszóma-mutációja kialakulhat mind a szülők hasonló mutációinak hátterében, mind azok hiányában. Az Angelman-szindróma (AS) örökletes jellege nem egyértelműen meghatározott, de a kromoszóma-mutációval rendelkező szülőknél a patológia kialakulásának valószínűsége meglehetősen magas.
Az is érdekes, hogy ha egy családban már van egy AS-ben szenvedő gyermek, akkor egy százalék az esélye annak, hogy egy második gyermek is ugyanezzel a rendellenességgel születik, még akkor is, ha a szülők egészségesek.
Még mindig nincsenek pontos statisztikák az Angelman-szindrómában szenvedő betegek számáról. Talán az ok a tünetek sokfélesége, amelyek bizonyos összetételben jelentkezhetnek, vagy hosszú ideig egyáltalán nem jelentkezhetnek. Feltételezik, hogy a betegség prevalenciája: 20 000 újszülöttre vetítve 1 gyermek. De ez a szám nagyon hozzávetőleges.
Okoz Angelman-szindróma
Az Angelman-szindróma egy kromoszóma-rendellenesség orvosi elnevezése, de korántsem az egyetlen. Az emberek babagyerek-szindrómának, boldog báb-szindrómának, Petruska-szindrómának és nevető baba-szindrómának nevezik. Mindenféle nevet találnak ki (néha akár magukra a betegekre és szüleikre nézve is sértőeket), de a betegség az betegség, függetlenül attól, hogy mennyire viccesen néz ki, és függetlenül attól, hogy mi az oka.
Az Angelman-szindróma kialakulásának okai, sok más genetikai patológiához hasonlóan, minden esetben az egyik kromoszóma vagy a kromoszómakészlet egészének szerkezetében fellépő zavarok. De a mi esetünkben az egész probléma a 15-ös kromoszómában rejlik, amely az anyától öröklődik. Vagyis az apai kromoszómának ebben az esetben nincsenek eltérései, de a női kromoszóma bizonyos mutációkon megy keresztül.
A kromoszóma-rendellenesség típusa szerint az Angelman-szindrómát kromoszóma-mutációnak minősítik. Az ilyen mutációk a következők:
- Egy deléció (egy bizonyos génkészletet tartalmazó kromoszómaszakasz hiánya; ha az egyik gén hiányzik, mikrodelécióról beszélünk), amely két törés és egy újraegyesülés eredménye, amikor az eredeti kromoszóma egy szakasza elvész.
- Duplikáció (egy extra szakasz jelenléte egy kromoszómában, amely egy meglévő másolata), ami a legtöbb esetben egy személy halálához, ritkábban meddőséghez vezet.
- Inverzió (a kromoszóma egyik szakaszának 180 fokkal történő megfordítása, azaz az ellenkező irányú elfordulása, majd a benne lévő gének ellentétes sorrendben helyezkednek el), amikor a kromoszóma törött végei az eredetitől eltérő sorrendben kapcsolódnak össze.
- Inszerció (ha a kromoszóma genetikai anyagának egy része nincs a helyén),
- transzlokáció (ha egy kromoszóma egy bizonyos szakasza egy másik kromoszómához kapcsolódik; egy ilyen mutáció kölcsönös lehet szakaszok elvesztése nélkül).
Egy gyanútlan anyától kapott mutált kromoszóma miatt a gyermek arra van ítélve, hogy rendellenességekkel szülessen. Az Angelman-szindróma leggyakoribb okának továbbra is az anyai 15. kromoszóma delécióját tekintik, amikor egy kis szakasz hiányzik. A "nevető baba" szindrómában a ritkább mutációk a következők:
- transzlokáció
- unipaternális disómia (ha a gyermek egy pár kromoszómát kapott az apától, az anyai kromoszóma hiányzik),
- a DNS-ben lévő gének mutációja, amelyek egyrészt a fő építőanyag (genetikai anyag), másrészt pedig a helyes használatára vonatkozó utasítások (különösen az ube3a gén mutációja az anyai kromoszómában).
Ezen mutációk egyikének jelenléte a szülőkben kockázati tényező az Angelman-szindróma kialakulásában gyermekeknél. De nemcsak a kromoszóma-mutációk, hanem a genomiális mutációk is (amelyek a kromoszóma-készletek mennyiségi változásával járnak, és gyakoribbak, mint a kromoszóma-mutációk) provokálhatják a betegség kialakulását egy gyermeknél. A gyakori genomiális mutációk közé tartozik a kromoszóma-triszómia (ha egy személy kromoszóma-készlete több mint 46 kromoszómát tartalmaz).
Ahhoz, hogy egy patológia megjelenjen egy gyermeknél, egyáltalán nem szükséges, hogy a szülőknek kromoszóma-rendellenességeik legyenek. Mégis van egy bizonyos százaléka a betegeknek, akiknek a betegsége örökletes.
Pathogenezis
Merüljünk el egy kicsit mélyebben a biológiában, pontosabban a genetikában. Minden egyes emberi szervezet genetikai információját 23 kromoszómapár tartalmazza. Egy pár egyik kromoszómája az apától, a másik az anyától öröklődik a gyermekre. Minden kromoszómapár alakjában és méretében különbözik, és bizonyos információkat hordoz. Így a 23. kromoszómapár (X és Y kromoszómák) felelős a baba nemi jellegeinek kialakulásáért (XX - lány, XY - fiú, míg az Y kromoszómát csak az apától kaphatja meg a gyermek).
Ideális esetben a gyermek 46 kromoszómát kap a szüleitől, amelyek meghatározzák genetikai jellemzőit, előre meghatározva őt egyénként. A nagyobb kromoszómaszámot triszómiának nevezzük, és a normától való eltérésnek tekintjük. Például a 47-es kromoszóma jelenléte a kromoszómakészletben (kariotípus, amely meghatározza a fajt és az egyéni jellemzőket) okozza a Down-szindróma előfordulását.
Ha a kromoszómákat speciális festékkel festjük, akkor mikroszkóp alatt különböző árnyalatú csíkokat láthatunk mindegyik mentén. Minden csíkon belül hatalmas számú gén található. Ezeket a csíkokat a tudósok számozták, és rögzített helyen vannak. Az egyik csík hiánya eltérésnek tekinthető a normától. Angelman-szindrómában nagyon gyakran megfigyelhető az anyai kromoszóma q11-q13 intervallumában található szegmensek hiánya, amelyek a hosszú karban helyezkednek el, és amelyekben a DNS-bázisok száma mindössze 4 millió.
A kromoszóma fő alkotóeleme egy hihetetlenül hosszú DNS-molekula, amely több ezer gént és több tíz- és százmillió nitrogénbázist tartalmaz. Így a 15-ös kromoszóma, amely az Angelman-szindróma és számos más betegség kialakulásáért felelős, 1200 gént és körülbelül 100 millió bázist tartalmaz. A DNS-molekula szerkezetében bekövetkező bármilyen zavar minden bizonnyal befolyásolja a születendő gyermek megjelenését és fejlődését.
A génekben található genetikai információ fehérjévé vagy RNS-sé alakul. Ezt a folyamatot génexpressziónak nevezzük. Ily módon a szülőktől kapott genetikai információ formát és tartalmat is kap, amely egyedi női vagy férfi örökösükben testesül meg.
Számos nem klasszikus öröklési típusú patológia létezik, beleértve az Angelman-szindrómát is, amelyben a szülőktől párosított kromoszómák részeként kapott gének a szülők egyedi lenyomatát viselik, és különböző módon nyilvánulnak meg.
Tehát az Angelman-szindróma a genomikus imprinting feltűnő példája, amelyben a gyermek testében a génexpresszió közvetlenül függ attól, hogy melyik szülőtől kapták az allélokat (egy gén különböző formái, amelyeket az apától és az anyától kaptak, a párosított kromoszómák azonos szakaszain helyezkednek el). Vagyis csak az anyai kromoszóma rendellenességei vezetnek a szindróma kialakulásához, míg az apai kromoszóma mutációi és szerkezeti rendellenességei teljesen más patológiákat okoznak.
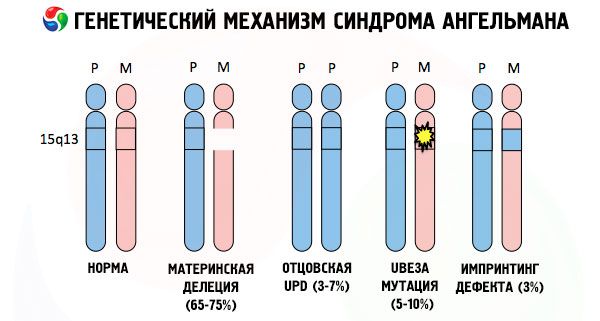
Ebben a patológiában bizonyos gének hiányoznak az anyai kromoszómában, vagy egyes gének aktivitása csökken/elvész (az esetek túlnyomó többségében az ube3a géné, amely az ubiquitin, egy más fehérjék lebontását szabályozó fehérje anyagcseréjében vesz részt). Ennek eredményeként a gyermeknél mentális fejlődési rendellenességeket és fizikai deformitásokat diagnosztizálnak.
Tünetek Angelman-szindróma
Az Angelman-szindróma tünetei a gyermek életének és fejlődésének különböző aspektusait befolyásolják: fizikai, neurológiai, mentális. Ennek alapján 3 tünetcsoport azonosítható, amelyek a patológia kialakulására utalnak.
- Külső vagy fizikai tünetek:
- aránytalanul kicsi fej a normál méretű testhez és végtagokhoz képest,
- túl széles száj,
- szinte mindig mosoly van az arcán (nyitott szájjal),
- ritka fogak,
- keskeny felső ajak,
- gyakran kiálló széles nyelv,
- kiálló alsó állkapocs,
- hegyes áll,
- nagyon világos bőr, gyakran szőrzet (albinizmus, ami azzal a ténnyel jár, hogy a szervezet nem termel melanin pigmentet),
- sötét foltok a világos bőrön (hipopigmentáció a melanin elégtelen termelése miatt)
- fizikai vagy külső tünetek: szembetegségek, például kancsalság vagy látóideg-sorvadás,
- gerincferdülés (szkoliózis),
- merev lábak (járás közben az ember nem hajlítja be a térdét az ízületek alacsony mozgékonysága miatt, ezért hasonlítják össze egy baba járásával).
- A mentális és érzelmi fejlődéssel kapcsolatos tünetek:
- súlyos mentális retardáció,
- túlzottan érzelmes, zajos, nyűgös viselkedés,
- gyakori tapsolás,
- kifejezett barátságosság, amelyet az arcán lévő állandó mosoly hangsúlyoz,
- gyakori nevetés ok nélkül.
- Neurológiai tünetek:
- végtagok remegése,
- a mozgások elégtelen koordinációja egyensúlyvesztéssel,
- csökkent izomtónus,
- különféle alvászavarok,
- gyakori hisztériás rohamok gyermekkorban,
- beszédzavarok (a gyermek későn kezd el beszélni, gyenge a kommunikációs készsége és elmosódott a beszéde),
- hiperaktivitás a fokozott ingerlékenység hátterében,
- koncentrációs és tanulási nehézségek.
De ez a betegség általánosított képe. Valójában az Angelman-szindróma klinikai képe nagymértékben függ a betegség fejlődési stádiumától és a patológiát okozó kromoszóma-mutáció típusától. Ez azt jelenti, hogy a betegség tünetei jelentősen eltérhetnek a különböző betegeknél, ami sokáig nem tette lehetővé, hogy megkülönböztessük a patológiát a hasonló klinikai képpel rendelkezőktől.
A tünetek teljes száma közül kiemelhetjük azokat, amelyek kivétel nélkül minden betegre jellemzőek:
- súlyos mentális retardáció,
- nem megfelelő viselkedés (indokolatlan nevetés, fokozott ingerlékenység, koncentrációzavar, eufória),
- a motoros készségek fejletlensége,
- a mozgások rossz koordinációja, járási ataxia (egyenetlen tempó, oldalirányú imbolygás stb.), végtagok remegése.
- beszédfejlődési zavar, amelyben a nem verbális kommunikációs eszközök dominálnak.
A betegek túlnyomó többségénél tapasztalt tünetek közül a következőket lehet megkülönböztetni:
- a fej és a test közötti aránytalanság, amelyet a fizikai fejlődés késése okoz,
- sok betegnél a koponya alakja olyan, hogy az agy mérete kisebb marad, mint az egészséges embereknél (mikrokefália),
- 3 éves kor előtti epilepsziás rohamok, amelyek erőssége és gyakorisága idősebb korban fokozatosan csökken,
- az EEG-paraméterek torzulása (alacsony frekvenciájú hullámok ingadozása és nagy amplitúdója).
Ezek a tünetek meglehetősen gyakoriak, azonban az Angelman-szindrómában szenvedő betegek 20%-ánál nem jelentkeznek.
Még ritkábban lehetséges a betegség ilyen megnyilvánulásainak diagnosztizálása, mint:
- súlyos vagy enyhe kancsalság,
- a nyelv mozgásának gyenge kontrollja, aminek következtében a betegek gyakran ok nélkül kinyújtják a nyelvüket,
- nyelési és szopási nehézségek, különösen kisgyermekeknél,
- a bőr és a szem pigmentációjának zavara,
- járás közben felemelt vagy behajlított karokkal,
- hiperreflexia,
- alvászavarok, különösen gyermekkorban,
- gyakori nyálfolyás,
- kielégíthetetlen szomjúság,
- túlzottan aktív rágási mozgások,
- hőérzékenység,
- lapos fej hátulja,
- kiálló alsó állkapocs,
- sima tenyerek.
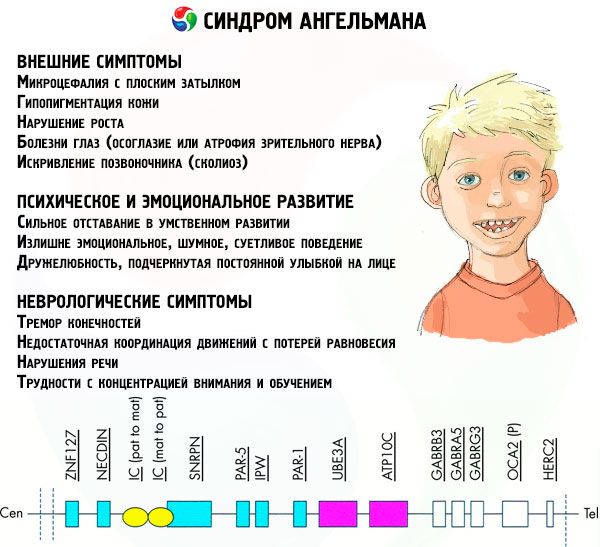
A betegek meglehetősen nagy százalékánál vizelési problémák jelentkeznek, melyeket rosszul kontrollálnak, a finommotoros készségek károsodása, ami nehézségeket okoz az önellátásban és a tanulásban, valamint a túlsúly. Szinte minden beteg később éli meg a pubertást, mint egészséges társai.
Az Angelman-szindrómás gyermekek jól érzékelik és megértik a szóbeli beszédet, de nem akarnak részt venni a beszélgetésben, beszédüket a mindennapi életben szükséges több tucat szóra korlátozzák. Felnőttkorban azonban ezek a betegek fiatalabbnak tűnnek, mint genetikai patológiák nélküli társaik.
Az Angelman-szindróma számos tünete változó, így a betegség klinikai képe az életkorral jelentősen változik. A görcsök és az epilepsziás rohamok ritkábbak vagy teljesen eltűnnek, a beteg kevésbé izgatottá válik, és az alvás javul.
Komplikációk és következmények
Az Angelman-szindróma egy súlyos, jelenleg gyakorlatilag gyógyíthatatlan kromoszóma-rendellenesség, amely megfosztja a betegeket a normális élet lehetőségétől. Az, hogy milyen lesz egy AS-ben szenvedő gyermek élete, nagymértékben függ a kromoszóma-rendellenesség típusától.
Egy kromoszómaszegmens megkettőződése a legtöbb esetben összeegyeztethetetlen az élettel. És még ha az ilyen betegek nem is halnak meg csecsemőkorban és nem érik el a pubertáskort, nincs esélyük gyermekvállalásra.
Az Angelman-szindrómában leggyakrabban előforduló gének egy részének deléciója vagy hiánya akadályozza a gyermek járás- és beszédtanulását. Az ilyen gyermekeknél súlyosabb a mentális retardáció, és az epilepsziás rohamok gyakrabban fordulnak elő, intenzitásuk pedig sokkal nagyobb, mint más kromoszóma-rendellenességekkel küzdő betegeknél.
Ha csak egyetlen gén mutációja van, kellő figyelemmel és megközelítéssel a gyermek megtanítható az öngondoskodás, a kommunikáció és az interakció alapjaira egy csoportban, bár a fejlődésben továbbra is lemarad társai mögött.
Az Angelman-szindrómás, természetüknél fogva kedves gyermekek számára a legfontosabb a szülők szeretete és figyelme. Csak ebben az esetben fog a gyermek oktatása gyümölcsöt hozni, még ha csekély mértékben is. Természetesen az AS-ben szenvedő betegek nem tudnak majd rendes iskolában tanulni. Speciális órákra van szükségük, ahol a gyerekeket először a koncentrációra tanítják, majd fokozatosan elsajátítják az iskolai ismeretek alapjait.
Diagnostics Angelman-szindróma
Az Angelman-szindróma egy veleszületett fejlődési rendellenesség. Bizonyos körülmények miatt azonban gyakran lehetetlen csecsemő- és kisgyermekkorban diagnosztizálni. Ez a csecsemők és 3 év alatti gyermekek tüneteinek nem specifikusságával és gyenge kifejeződésével magyarázható. Ráadásul a betegség előfordulása hazánkban nem olyan nagy, hogy az orvosok megtanulták volna felismerni a társaik között.
Az Angelman-szindróma csecsemőknél csökkent izomtónusként jelentkezhet, ami táplálkozási problémákban (a szopási és nyelési reflex gyengesége), később pedig a járástanulás nehézségeiben nyilvánul meg (az ilyen gyermekek sokkal később kezdenek járni). Ezek a tünetek a baba fejlődési rendellenességének első jelei, amely könnyen összefüggésben lehet egy kromoszóma-rendellenességgel. Csak genetikai elemzés erősítheti meg ezt a feltételezést.
Különös figyelmet fordítanak azokra a gyermekekre, akiknek a szülei különféle genomikai vagy kromoszóma-rendellenességekkel küzdenek. Végül is a betegség eleinte nem feltétlenül jelentkezik, és ha időben felismerik a patológiát, a gyermekkel való intenzív munka megkezdésével jelentősen nagyobb sikereket lehet elérni a tanulásban, lassítva a betegség progresszióját.
Ha a szülőknek különféle kromoszóma-rendellenességeik vannak, a genetikai elemzést még a baba születése előtt elvégzik, mivel az SA az egyik olyan patológia, amely már az embrionális stádiumban kimutatható.
A genetikai kutatáshoz szükséges anyagok gyűjtése kétféleképpen történhet:
- invazív (bizonyos kockázati százalékkal, mivel a méhbe kell behatolni a magzatvíz mintájának vételéhez),
- nem invazív (a baba DNS-ének elemzése az anya véréből).
Ezután a következő tanulmányokat végzik el:
- fluoreszcens in situ hibridizáció (FISH-módszer) – egy speciális festékkel jelölt DNS-próba kötődése a vizsgált DNS-hez, majd mikroszkópos vizsgálat.
- az ube3a gén és az imprintált gének mutációinak elemzése,
- DNS-metilációs analízis a genetikában alkalmazott speciális módszerekkel.
A genetikai tesztek meglehetősen pontos információkat nyújtanak kromoszóma-rendellenességek esetén, ami azt jelenti, hogy a leendő szülők előre tudják, mire kell felkészülniük. Vannak azonban kivételek. A betegek egy bizonyos csoportjánál, minden patológiára utaló tünet jelenlétében a teszteredmények normálisak maradnak. Vagyis a patológia csak a gyermek kisgyermekkorától kezdve történő gondos megfigyelésével azonosítható: hogyan eszik, mikor kezdett járni és beszélni, behajlítja-e a lábát járás közben stb.
A FISH módszer mellett az Angelman-szindróma instrumentális diagnosztikai módszerei közül kiemelhető a tomográfia (CT vagy MRI), amely segít meghatározni az agy állapotát és méretét, valamint az elektroencefalogram (EEG), amely megmutatja, hogyan működnek az agy egyes részei.
Az orvosok általában 3-7 éves korban állítanak fel végleges diagnózist, amikor a betegnek már a legtöbb tünete van, és a betegség fejlődésének dinamikája látható.
Milyen tesztekre van szükség?
Megkülönböztető diagnózis
Az Angelman-szindróma egy genetikai patológia, amelynek gyakorlatilag nincsenek specifikus tünetei. A legtöbb tünet egyaránt utalhat mind az Angelman-szindrómára, mind más genetikai patológiákra.
Az Angelman-szindróma differenciáldiagnózisát a következő patológiákkal végzik:
- Pitt-Hopkins szindróma (a betegekre jellemző a mentális retardáció, a vidám természet, a mosolygás, a szájuk meglehetősen nagy és széles, mikrokefália is megfigyelhető). A különbség a hiperventiláció és a légzésvisszatartás rohamai ébrenléti állapotban.
- Christianson-szindróma (a betegek szellemileg visszamaradott, vidám természetű, beszédképtelen emberek, akiket mikrocefália, ataxia, görcsök, akaratlan izommozgások jellemeznek).
- Mowat-Wilson szindróma (tünetek: mentális retardáció, epilepsziás rohamok, hegyes áll, nyitott száj, vidám arckifejezés, mikrokefália). Jellemzők: nagy távolság a szemek között, befelé dőlő szemek, lekerekített orrhegy, hátrafelé ívelt fülkagyló.
- Kabuki-szindróma (enyhe vagy közepes fokú mentális retardáció, beszéd- és motoros problémák, izomgyengeség, epilepsziás rohamok, kisfejűség, hosszú viszketésközi szünetek és koordinációs zavarok jellemzik). Ívelt szemöldök, az alsó szemhéj kifelé dőlő oldalsó része, távol ülő szemek, hosszú szemhéjhasadékok hosszú, vastag szempillákkal.
- Rett-szindróma (az AS-tól való megkülönböztetés nőknél). Tünetek: megkésett beszédfejlődés, görcsrohamok, mikrokefália. A különbség az, hogy nincs vidám kifejezés az arcon, apnoe és apraxia rohamok jelentkeznek, amelyek idővel súlyosbodnak.
- Autoszomális recesszíven öröklődő mentális lassulás szindróma 38 (tünetek: kifejezett mentális retardáció a motoros készségek és a beszéd késésével, izomgyengeség, táplálkozási problémák csecsemőkorban, impulzivitás). Megkülönböztető jellemzője az írisz kék színe.
- MECP 2 génduplikációs szindróma (férfiaknál a SA-tól való differenciálódás). Tünetek: súlyos mentális retardáció, gyermekkor óta fennálló izomgyengeség, beszédproblémák vagy beszédképtelenség, epilepszia. Különbségek: progresszív myopathia, folyamatosan visszatérő fertőzések.
- Kleefstra-szindróma (tünetek: beszéd- és gondolkodási problémák, izomgyengeség, alvászavarok, figyelemhiány, nyitott száj, hiperaktivitás, görcsrohamok, ataxia, egyensúlyzavarok). Jellemzők: lapos arc, rövid, pisze orr, távol ülő szemek, nagy, kifelé ívelő alsó ajak, agresszív kitörések.
- Smith-Magenis szindróma (görcsrohamok, alvászavarok, intellektuális és motoros fejlődési zavarok jellemzik). Jellemző jellemzői a széles és lapos arc, valamint a kiemelkedő homlok.
- Koolen-de-Vries szindróma (enyhe vagy közepes fokú mentális retardáció, izomgyengeség, görcsrohamok, barátságosság). Megkülönböztető jellemzők: hosszú arc magas homlokkal, kiálló fülek, ferde szemek, nagy ízületi mozgékonyság, veleszületett szívhibák.
- Phelan-McDermid szindróma (tünetek: mentális retardáció, beszédzavarok vagy beszéd hiánya). Jellemzők: nagy, fejlett izmokkal rendelkező kezek, születéstől fogva fennálló izomgyengeség, gyenge izzadás.
Az olyan patológiák, mint az adenil-szukcinát-hiány, az autoszomális recesszív mentális retardáció szindróma 1, a 2q23.1 kromoszóma duplikációs szindróma, a FOXG1, STXBP1 vagy MEF2C gén haploinsufficiency szindrómák és néhány más, az Angelman-szindrómához hasonló tünetekkel „büszkélkedhetnek”.
Az orvos feladata a pontos diagnózis felállítása, az Angelman-szindróma megkülönböztetése a hasonló tünetekkel járó patológiáktól, és hatékony kezelés felírása, amely releváns a betegség diagnosztizált stádiumához.
Ki kapcsolódni?
Kezelés Angelman-szindróma
Az Angelman-szindróma egyike azoknak a patológiáknak, amelyekre az orvostudomány még mindig keresi a hatékony kezelést. A betegség etiológiai kezelése különféle módszerek és eszközök fejlesztése folyamatban van, amelyek közül sokat még nem teszteltek embereken. Ez azt jelenti, hogy az orvosoknak egyelőre a tüneti terápiára kell korlátozniuk magukat, amely valamilyen módon enyhíti a marionett-szindrómában szenvedő gyermekek és felnőttek irigylésre méltó helyzetét, akik epilepsziás rohamoktól, nyálfolyástól, alacsony vérnyomástól és alvászavaroktól szenvednek.
Így egy megfelelően kiválasztott görcsgátló gyógyszer segítségével csökkenthető az epilepsziás rohamok gyakorisága és erőssége. A nehézség azonban abban rejlik, hogy az SA-ban szenvedő betegek rohamai abban különböznek a szokásos epilepsziás rohamoktól, hogy többféle roham jellemzi őket, ami azt jelenti, hogy az állapot enyhíthető több gyógyszer egyidejű adagolásával.
Az Angelman-szindróma kezelésére használt legnépszerűbb görcsgátlók a valproinsav, a topiramát, a lamotrigin, a levetiracetám, a klonazepám és az ezeken alapuló gyógyszerek. Kevésbé elterjedtek a karmazepin, fenitoin, fenobarbitál és etoszuximid alapú gyógyszerek, mivel ezek némelyike paradox hatást válthat ki, amely az epilepsziás rohamok felerősödésében és gyakoriságának növelésében rejlik. Ez akkor fordul elő, ha a gyógyszert monoterápiaként alkalmazzák.
A nyáladzás kezelésére általában két módszert alkalmaznak: gyógyszeres (a nyáltermelést csökkentő gyógyszerek) és sebészeti beavatkozást, amely a nyálcsatornák újbóli beültetését jelenti. Az SA esetében azonban ezek a módszerek hatástalannak minősülnek, és a kérdés továbbra is nyitott marad. A szülőknek és az ilyen betegeket gondozóknak különös figyelmet kell fordítaniuk erre a kérdésre, mivel maguk a betegek általában nem tudják kontrollálni a nyáladzást, és néhányan egyszerűen nem tudnak gondoskodni magukról.
Egy másik probléma a rövid alvásidő. Az Angelman-szindrómás gyermekek gyakran nem alszanak többet 5 óránál, ami negatívan befolyásolja az egész test működését. A könnyen izgalomba jönő, aktív gyerekek, akik szeretik a játékokat és a kommunikációt (még akkor is, ha megpróbálják a nonverbális módszerekre korlátozni magukat), napközben észrevehetően elfáradnak. A jó pihenéshez a testnek mély, teljes alvásra van szüksége, de pontosan ez a csapda.
Úgy tűnik, hogy az idegrendszert nyugtató nyugtató gyógyszerek (fenotiazinok és atipikus antipszichotikumok) elegendőek kellene, hogy legyenek az ingerlékeny betegek alvásának javítására. Az AS esetében azonban az ilyen gyógyszerek alkalmazása negatív hatásokkal jár. Ezért az orvosok továbbra is az enyhe altatókat részesítik előnyben, mint például a melatonin (egy természetes hormonális gyógyszer, amely az alváshormonon alapul), amelyet a betegeknek lefekvés előtt egy órával adnak be 1 tabletta mennyiségben, és a difenhidramin. Az adagolás gyakoriságát és az adagolást az orvos határozza meg a beteg állapotától és életkorától függően.
Az Angelman-szindrómában szenvedő betegeknek néha emésztési és székletproblémáik vannak. A széklet állapotát hashajtókkal (lehetőleg gyógynövényesekkel) javíthatja.
Vagy másképp is megközelítheti a problémát, ahogy az amerikai orvosok tették, az autizmus kezelésének egyes módszereire alapozva, mivel az AS-re jellemző számos tünet az autizmusra is jellemző (impulzivitás, akaratlan mozgások, ismétlődő cselekvések, figyelemhiány, kommunikációs problémák stb.). Megjegyezték, hogy a szekretin hormon bevezetése, amely normalizálja az emésztést és a székletürítést, pozitív hatással van a betegek figyelmére, az oxitocin pedig segít javítani a gyermek kognitív képességeit és memóriáját, valamint a helyes viselkedést.
Igaz, a hormonok önmagukban nem elegendőek, különösen, ha gyermekekről van szó. Angelman-szindróma esetén viselkedésterápia, pszichológussal és logopédussal való munka (nonverbális kommunikációs módszerek és jelnyelv oktatása) javasolt. Az ilyen gyermekek oktatásának egyéni programon kell alapulnia, speciálisan képzett tanárok, pszichológus és szülők részvételével. Sajnos ez nem mindenhol lehetséges, és a családok magukra maradnak a problémájukkal.
Mivel sok fiatal AS-beteg alacsony izomtónustól és ízületi problémáktól szenved, nagy figyelmet fordítanak a fizioterápiára. Az orvosok leggyakrabban paraffinalkalmazásokat, elektroforézist és mágnesterápiát alkalmaznak.
Az aktív tonizáló masszázs és a terápiás fizikoedzés speciális gyakorlatai segítenek a beteg gyermeknek egy idő után talpra állni és magabiztosan járni. Különösen hasznos ebből a szempontból az aquagimnasztika, amelyet hűvös vízben történő alváshoz ajánlanak. Növeli az izomtónust, és megtanítja a gyermeket a teste irányítására és a mozgások koordinálására.
Görcsgátló kezelés
Az Angelman-szindróma legveszélyesebb tünete az epilepsziához hasonló rohamok. Ez a tünet a betegek 80%-ánál figyelhető meg, ami azt jelenti, hogy mindegyiküknek hatékony görcsoldó kezelést kell felírnia.
Az epilepsziás rohamok kezelését vitaminok és görcsoldók segítségével végzik. Angelman-szindróma esetén, görcsös szindrómával együtt, a B-vitaminok, valamint a C-, D- és E-vitaminok hasznosak lesznek. De a vitaminterápia önálló felírása ebben az esetben nagyon veszélyes, mivel a vitaminok ellenőrizetlen bevitele csökkentheti az antiepileptikumok hatékonyságát, és új, súlyosabb és elhúzódóbb rohamokat válthat ki.
A görcsgátló gyógyszerek kiválasztását és hatékony adagolásuk felírását szintén szakorvosnak kell elvégeznie. Ő dönti el azt is, hogy egy gyógyszer elegendő lesz-e, vagy a betegnek hosszú ideig 2 vagy több gyógyszert kell szednie.
A legtöbb beteg számára az orvosok valproinsav tartalmú gyógyszereket (Valproinsav, Depakine, Convulex, Valparin stb.) írnak fel, amelyek megakadályozzák a rohamokat, javítják a betegek hangulatát és mentális állapotát.
A valproinsav tabletták, szirup és injekciós oldatok formájában kapható. A legnépszerűbb gyógyszer a "Depakine" nevű retard gyógyszer tablettákban és intravénás beadásra szánt oldat formájában. A gyógyszer adagját az orvos határozza meg egyénileg, a beteg súlyától, életkorától és állapotától függően.
A gyógyszert naponta 2-3 alkalommal, étkezés közben kell bevenni. Az átlagos napi adag 20-30 mg testsúlykilogrammonként, a maximális adag 50 mg/kg naponta.
Ellenjavallatok. Ne alkalmazza máj- és hasnyálmirigy-működési zavar, vérzéses diatézis, hepatitisz, porfiria és a gyógyszerrel szembeni túlérzékenység esetén.
A mellékhatások közé tartozik a kézremegés, az emésztési és székletzavarok, valamint a testsúlyváltozások.
A "topiramát" szintén a választott gyógyszer az SA számára. Tabletta formájában kapható, és mind monoterápia részeként, mind más gyógyszerekkel kombinálva alkalmazzák.
Alkalmazás módja és adagolás. A tablettákat szájon át, étkezéstől függetlenül kell bevenni. A kezdeti adag felnőtteknek napi 25-50 mg, gyermekeknek 0,5-1 mg/testtömegkg. Az adagot hetente az orvos utasítása szerint emelik.
A gyógyszert nem szabad terhesség és szoptatás alatt, valamint az összetevőivel szembeni túlérzékenység esetén szedni. A gyógyszernek számos különböző mellékhatása van.
Angelman-szindróma esetén felírható gyógyszerek: Clomazepam, Rivotril, Lamotrigin, Seizar, Lamictal, Levetiracetam, Keppra, Epiterra stb.
 [ 16 ], [ 17 ], [ 18 ], [ 19 ]
[ 16 ], [ 17 ], [ 18 ], [ 19 ]
Hagyományos orvoslás és homeopátia
A hagyományos orvoslás, mint például a homeopátiás készítmények, természetesen viszonylag biztonságosak, de az Angelman-szindróma ilyen kezelésének hatékonysága ellentmondásosnak tekinthető.
Bár a népi gyógymódok is segíthetnek bizonyos dolgokban. Epilepsziás rohamok megállításáról beszélünk. Ebben a tekintetben a gyógynövényes kezelés meglehetősen hatékony lehet.
Jó hatást fejt ki a bazsarózsa, édesgyökér és békalencse alapú gyógygyűjtemény (az összetevőket egyenlő mennyiségben kell bevenni). A gyógynövényeket lisztté kell őrölni. A szedés kezdetétől számított 2 hét elteltével a rohamok gyakoriságának jelentős csökkenését tapasztalhatja.
Görcsök esetén levendulafőzet (1 teáskanálnyi egy pohár forrásban lévő vízhez) is hasznos. A keveréket 5 percig forraljuk, majd fél órán át áztatjuk. A gyógyszert éjszaka 14 napig szedjük.
Az anyafű vizes (vagy alkoholos) infúzióját hatékonynak tekintik epilepsziás rohamok esetén.
Az Angelman-szindróma rohamainak megelőzésére szolgáló homeopátiás készítmények közül használhatunk kamilla és anyagyökér, Acidum hydrocyanicum, Argentum nitricum, Kalium bromatum, Arsenicum album alapú gyógyszereket. De figyelembe kell venni, hogy csak egy homeopátiás orvos írhat fel hatékony és biztonságos gyógyszeradagot minden egyes esetben.
Megelőzés
Ahogy az olvasó valószínűleg már megértette, az orvostudomány egyelőre nem képes megelőzni a génmutációkat és más kromoszóma-rendellenességeket, valamint korrigálni a helyzetet. Ez bárkivel megtörténhet, mivel az Angelman-szindrómás gyermekek egészséges szülőktől születnek, és a genetika, amely jelenleg az orvostudomány egyik legkevésbé kutatott ága, erre még nem tud magyarázatot adni.
Az egyetlen dolog, amit tehetünk, az, hogy felelősségteljesen közelítsük meg a terhességtervezést, időben regisztráljunk és részt vegyünk vizsgálatokon. De ismét, egy ilyen intézkedés inkább oktató jellegű lesz, mint megelőző, mint bármely vizsgálat. A fiatal szülők azonban előre tudják, mire kell felkészülniük, és pozitív válasz esetén eldöntik, hogy vállalhatnak-e olyan felelősséget, mint egy beteg gyermek nevelése.
Előrejelzés
Az Angelman-szindróma prognózisa a kromoszóma-rendellenesség jellegétől és felismerésének időszerűségétől függ. A legsúlyosabban azokat a gyermekeket érinti, akiknek a 15-ös kromoszómáján "gének" hiányosságai (deléció) vannak. Az ilyen betegek járási és beszédképességének valószínűsége rendkívül alacsony. Más esetek gondos megközelítéssel és a gyermek iránti szeretettel korrigálhatók.
Sajnos az ilyen betegek nem lesznek képesek a társadalom teljes értékű tagjaivá válni, annak ellenére, hogy korántsem ostobák, értik a beszédet és annak jelentését. Kommunikációs problémáik azonban életük végéig megmaradnak. A betegeket már gyermekkoruktól fogva meg lehet tanítani a jelnyelvre, de nem lehet őket szavakkal kommunikálni. A „beszélő” betegek szókincse a mindennapi életben használt szavak minimumára korlátozódik (5-15 szó).
Ami az Angelman-szindrómás betegek várható élettartamát és általános egészségi állapotát illeti, az itt látható számok az átlagos értékek körül ingadoznak. Felnőttkorban a betegek többnyire olyan egészségügyi problémákkal szembesülnek, mint a gerincferdülés és az elhízás, amelyek a megfelelő kezelési megközelítéssel nem életveszélyesek.