A cikk orvosi szakértője

Új kiadványok
Angelmann-szindróma gyermekeknél és felnőtteknél
Utolsó ellenőrzés: 23.04.2024

Minden iLive-tartalmat orvosi szempontból felülvizsgáltak vagy tényszerűen ellenőriznek, hogy a lehető legtöbb tényszerű pontosságot biztosítsák.
Szigorú beszerzési iránymutatásunk van, és csak a jó hírű média oldalakhoz, az akadémiai kutatóintézetekhez és, ha lehetséges, orvosilag felülvizsgált tanulmányokhoz kapcsolódik. Ne feledje, hogy a zárójelben ([1], [2] stb.) Szereplő számok ezekre a tanulmányokra kattintható linkek.
Ha úgy érzi, hogy a tartalom bármely pontatlan, elavult vagy más módon megkérdőjelezhető, jelölje ki, és nyomja meg a Ctrl + Enter billentyűt.

Számos olyan betegség létezik, amelyekben olyan kifejezések, mint a "vigyázz magadra, és nem betegedtek", legalább nevetségesek. Ez a kórtan, amelyben néhány mentális és fizikai rendellenesség még a születés előtt beágyazódik a gyermek testébe, de a szülőknek nincs bűnössége. Az ilyen betegségeket a kromoszómakészletekben mutációk vagy rendellenességek okozzák, és kromoszómális vagy genetikai jellegűek. Angelman-szindróma, Down-szindróma, Patau, Edwards, Turner, Prader-Willi csak egy része a genetikai betegségeknek egy meglehetősen tisztességes listából.
Egy boldog ember szindróma
Ezúttal beszélünk a betegség, melynek névadója a brit gyermekorvos Harry Angelman az elsőként felvetette a problémát 1965 év, szemben a estéjén gyakorlat három szokatlan gyermek, egyesült közös sajátos tüneteket. Az orvos ezeket a gyerekeket bábszínészeknek nevezte, és róluk írt róla egy cikket, amelyet eredetileg "Báb-gyerekeknek" nevezett. Maga a cikk és neve a Verona egyik múzeumában látható kép benyomása alapján íródott. A kép egy nevető fiút ábrázolt, és "Boy-báb" -nak nevezték. A képen ábrázolt gyermek együttese a három gyermekével, akivel Angelman egyszer találkozott a gyakorlatában, és arra ösztönözte a gyermekorvosát, hogy a meglévő betegsége miatt egy csoportba egyesítse a gyermekeket.
Nem meglepő, hogy a gyerekek észrevették a cikket, hogy más orvosok nem vették észre. Végtére is, első pillantásra úgy tűnt, hogy teljesen különböző betegségeik vannak, így a betegség általános klinikai képét három különböző eset különböztette meg. Az "új" kromoszómák kórokozója érdekes lehet más tudósok számára, de abban az időben a genetika még nem volt elég fejlett ahhoz, hogy megerősítse az angol orvos hipotézisét. Ezért a cikk egy bizonyos érdeklődés után régóta elhagyta a távoli ezredbe.
Az Angelman-szindróma következő megemlítése, és így Angliában, az angol gyermekgyógyásznak, a 20. Század nyolcvanas évének elejére nyúlik vissza. És csak 1987-ben sikerült megtalálni az oka annak, hogy a gyermekek egy kis része olyan eltérésekkel szülött, amelyek oldaláról folyamatosan mosolyognak és boldogok. Valójában ez nem így van, és a mosoly csak fintor, mögötte fekszik a boldogtalan emberi lélek és a szülők fájdalma.
Járványtan
A gyermekek kromoszómális mutációja - a statisztikák szerint - a szülők ilyen mutációinak hátterében is kialakulhat, és ilyen hiányában. Az Angelman-szindrómában (SA) nincs egyértelmű örökletes karakter, de a kromoszómális mutációkban szenvedő szülők patológiájának kialakulásának valószínűsége meglehetősen magas.
Érdekes, hogy ha egy családnak már van gyermeke egy SA-val, akkor van egy százalékos esélye arra, hogy ugyanolyan típusú második gyermeke legyen, még akkor is, ha a szülők egészségesek.
Az Anghelman-szindrómában szenvedő betegek számáról még mindig nincs pontos statisztika. Talán a hiba a különböző tünetek, amelyek egy bizonyos összetételben előfordulhatnak vagy sokáig nem merülnek fel. Feltételezzük, hogy a betegség előfordulási gyakorisága: 1 gyermek 20 000 újszülöttnél. De ez a szám nagyon közelítő.
Okoz angelman-szindróma
Az Angelmann-szindróma a kromoszóma patológiájának orvosi neve, de ez nem az egyetlen. Emberekben ez a betegség a báb gyermekek szindrómájának, a boldog báb szindrómájának és a Petrushka-szindrómának, valamint a nevető baba szindrómájának is nevezik. Igen, miféle nevek nem tudnak felidézni (néha sértik magukat a pácienseknek és a szüleiknek), de a betegség betegség, függetlenül attól, milyen furcsának tűnik kifelé és bármilyen oka lehet.
És az Angelman-szindróma kialakulásának okai, valamint számos más genetikai patológia, minden esetben az egyik kromoszóma vagy a kromoszóma egészének szerkezetében bekövetkezett megsértés. De csak a mi esetünkben az egész probléma az anyából továbbított 15 kromoszómában rejlik. Ie Ebben az esetben az apai kromoszómának nincsenek eltérései, de a nõnek bizonyos mutációk vannak.
A kromoszomális rendellenességek típusai szerint az Angelmann-szindróma kromoszómális mutációkra utal. Az ilyen mutációk:
- Törlése (hiánya kromoszóma tartalmazó régió egy adott génkészlet, és ha nem az egyik gén jön mikrodeléciók), amely az eredmény a két folytonossági és egy újraegyesítését, ha elveszett része az eredeti kromoszóma.
- Duplikáció (a kromoszómában lévő extra hely jelenléte, amely a már rendelkezésre álló másolat), amely a legtöbb esetben egy személy halálához vezet, ritkábban - a meddőséghez.
- Inversion (inverzió egy kromoszóma szakaszok 180 fok, azaz az ellenkező irányba, majd gének benne vannak elhelyezve fordított sorrendben), amikor a törött kromoszómavégek csatlakozik egy érdekében eltér az eredeti.
- Beillesztés (ha a genetikai anyag része a kromoszómában nincs a helyén),
- transzlokáció (ha a kromoszóma egy része csatlakozik egy másik kromoszómához, egy ilyen mutáció kölcsönös lehet a helyek elvesztése nélkül).
Egy mutáns kromoszómát egy gyanútlan anyától szerezve, a baba előre elszenvedett elvárásokkal született. Az Anghelman-szindróma kialakulásának leggyakoribb oka még mindig az anyai 15 kromoszóma törlése, amikor nincs benne kis terület. A "nevető baba" szindrómában kevésbé gyakori mutációk:
- transzlokáció,
- egy apai dioszómiát (ha a gyermek egy pár kromoszómát kap az apától, az anya kromoszóma hiányzik)
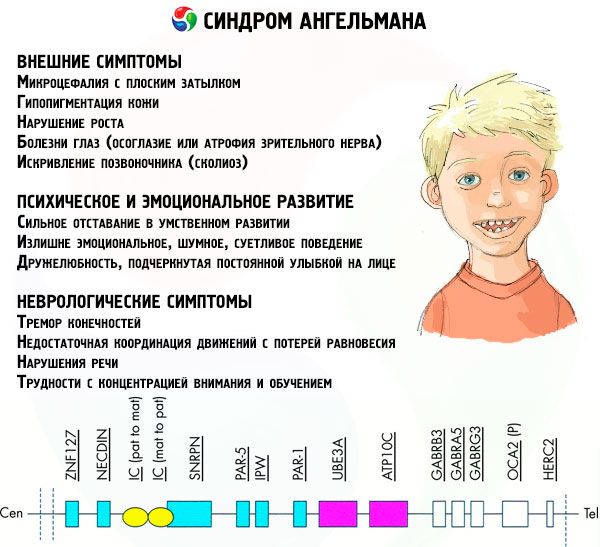
- a DNS-ben lévő gének mutációja, amelyek mind a fő építőanyag (genetikai) anyag, mind a helyes használatára vonatkozó utasítások (különösen az ube3a gén mutációja az anyai kromoszómában).
Az ilyen mutációk jelenléte a szülőkben kockázati tényező az Anghelman-szindrómában a gyermekeknél. De nemcsak a kromoszómális mutációk, hanem a genomiális mutációk (amelyek kromoszómakészletek mennyiségi változásával és gyakrabban megtalálhatók a kromoszómákban) a betegség kialakulásához vezethetnek a gyermekben. A közös genomiális mutációk a kromoszómák triszómájának tulajdoníthatók (ha egy személynek kromoszómakészlete több mint 46 kromoszómával rendelkezik).
A csecsemő patológiájához nem feltétlenül kell a szülőknek kromoszómális rendellenességek. És mégis vannak olyan betegek egy része, akiknek a betegsége örökletes.

Pathogenezis
Nézzünk egy kicsit a biológiában, pontosabban a genetikában. Minden egyes emberi test genetikai információját 23 pár kromoszóma tartalmazza. A pár egyik kromoszóma átadódik a gyermeknek az apa és a másik az anyából. Minden kromoszóma pár formában és méretben különbözik, és magában hordoz bizonyos információkat. Tehát, felelős megalakult a baba nemi jelleg 23 pár kromoszómát (X és Y kromoszómák) (XX - lány, fiú HY- az Y-kromoszómát, a baba kaphat csak az apa).
Ideális esetben egy gyermek a szüleitől 46 kromoszómát kap, amelyek a genetikai tulajdonságait alkotják, előre meghatározott személyként. A kromoszómák nagyobb számát triszómiának nevezik, és eltérésnek tekintik a normát. Például 47 kromoszóma jelenléte a kromoszómakészletben (kariotípus, amely meghatározza a fajok és az egyéni jellemzők) okozza a Down-szindróma kialakulását.
Ha a kromoszómákat speciális festékkel színezik, akkor a mikroszkópban különböző színű sávok láthatók egymás mellett. Az egyes sávokon belül hatalmas számú gén található. Mindezeket a sávokat tudósok számozzák, és állandó helyük van. Az egyik sáv hiánya eltér a normától való eltérésnek. Amikor Angelman szindróma gyakran megfigyelni anyai kromoszóma szegmensek közötti Q11-Q13, található a hosszú karján, a számú DNS bázisok, hogy csak körülbelül 4 millió.
A kromoszóma fő összetevője hihetetlenül hosszú DNS-molekula, amely több ezer gént és tíz és több száz millió nitrogéntartalmat tartalmaz. Így az Angelman-szindróma kialakulásáért felelős 15 kromoszóma és még sok más, 1200 gént és 100 millió bázist tartalmaz. A DNS-molekula szerkezetének bármely megsértése szükségszerűen befolyásolja a születendő gyermek megjelenését és fejlődését.
A génben található genetikai információ átalakul fehérjékké vagy RNS-ből. Ezt a folyamatot génexpressziónak nevezik. Így a szülõktõl kapott genetikai információ mind formát, mind tartalmat kap, amelyet a nõi vagy a férfi nemi örököse hordoz.
Van számos patológiás egy nonclassical típusú öröklés, beleértve Angelman-szindróma, amelyben a gének kapott szülők részeként a párosított kromoszómák egyedi lenyomata a szülők és megmutatkoznak a különböző módon.
Tehát, Angelman szindróma egy kiváló példa genomiális imprinting, miáltal az expressziós gének a szervezetben a gyermek közvetlenül függ akiktől szülő származó allélek (különböző formái ugyanazon gén nyert az apa és az anya található a azonos részek párosított kromoszómák) . Ie kialakulásához vezet egy szindróma rendellenességek a anyai kromoszómán, míg a mutációk és rendellenességek apai kromoszóma szerkezete oka nagyon különböző betegségek.
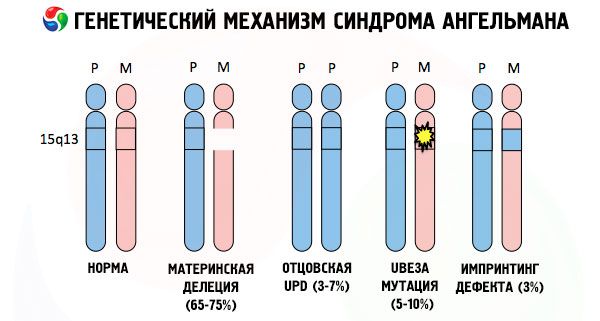
Ebben a betegségben hiányzik specifikus gének anyai kromoszóma vagy a veszteség / aktivitásának csökkenése az egyes gének (a legtöbb esetben UBE3A gén részt vesz az anyagcsere ubiquitin - fehérje lebomlását más szabályozó fehérjék). Ennek következtében a gyermeknek mentális fejlődési rendellenességek és fizikai deformitások diagnosztizáltak.
Tünetek angelman-szindróma
Az Angelman-szindróma tünetei a gyermek életének és fejlődésének különböző aspektusait érintik: fizikai, neurológiai és pszichikai jellegűek. Ennek alapján megkülönböztethetünk három tünetcsoportot, amelyek jelzik ezt a patológiát.
- Külső vagy fizikai tünetek:
- egy aránytalanul kis fej, a rendes méretű törzshez és végtagokhoz képest,
- túl széles száj,
- az arcon szinte mindig mosolyog (nyitott szájjal),
- ritka fogak,
- keskeny felső ajak,
- gyakran nyúlik ki széles nyelvet,
- kinyúló alsó állkapocs,
- éles áll,
- nagyon könnyű bőr, gyakran haj (albinizmus, azzal a ténnyel, hogy a szervezet nem termel pigment melanint),
- sötét foltok a könnyű bőrön (hypopigmentation miatt elégtelen melanin termelés)
- fizikai vagy külső tünetek: szembetegségek, például sztrabizmus vagy a látóideg sorvadása,
- a gerinc görbülete (scoliosis),
- merev lábak (amikor sétál, az ember nem térdel térdel az ízületek kis mozgása miatt, tehát összehasonlítva a bábjárással).
- A mentális és érzelmi fejlődéssel kapcsolatos tünetek:
- erős lemaradás a mentális fejlõdésben,
- feleslegesen érzelmes, zajos, könyörtelen viselkedés,
- gyakori tapsolás,
- az expressz barátságos, hangsúlyozva az állandó mosollyal az arcán,
- gyakori, ok nélküli nevetés.
- Neurológiai tünetek:
- végtagok tremorja,
- a mozgások elégtelen összehangolása, egyensúlyhiány,
- csökkent izomtónus,
- számos alvászavar,
- gyakori hisztériás rohamok gyermekkorban,
- beszédkárosodás (a gyermek későn beszél, rossz kommunikációs készséggel és elmosódott beszéddel),
- hiperaktivitás a fokozott izgatottsággal szemben,
- koncentrációs és képzési nehézségekkel.
De ez egy általános kép a betegségről. Tény, hogy az Anghelman-szindróma klinikai képe nagymértékben függ a betegség fejlődési stádiumától és a patológiát okozó kromoszómális mutáció típusától. Ez azt jelenti, hogy különböző betegeknél a betegség tünetei jelentősen eltérhetnek egymástól, ami hosszú időn keresztül nem engedte meg a betegség kóros elkülönítését hasonló klinikai képekkel.
A tünetek teljes számának azonosítása olyan, hogy kivétel nélkül minden beteg számára jellemző:
- súlyos eltérések a mentális fejlődésben,
- nem megfelelő magatartás (ok nélküli nevetés, fokozott izgatottság, rossz figyelemfelkeltés, eufóriaállapot),
- a motoros készségek elmaradása,
- a mozgások rossz koordinációja, a gyaloglás ataxia (egyenetlen ütem, oldalról oldalra ingadozás stb.), a végtagok remegése.
- a beszéd fejlesztésének megszegése a nem verbális kommunikációs eszközök túlsúlyával.
A betegek túlnyomó többségében előforduló tünetek közül megkülönböztethetünk ilyeneket:
- aránytalan fej és törzs, amelyet a fizikai fejlődés késleltetése okoz,
- sok esetben a koponya alakja olyan, hogy az agy mérete kisebb marad, mint az egészséges embereknél (mikrocefália),
- epilepsziás görcsrohamok legfeljebb 3 éves korban, az idősek fokozatos csökkenésével és gyakoriságával,
- az EEG mutatók torzulása (oszcillációk és az alacsony frekvenciájú hullámok nagy amplitúdója).
Ezek a tünetek gyakran fordulnak elő, azonban az Angelmann-szindrómában szenvedő betegek 20% -ánál hiányoznak.
Még ritkábban diagnosztizálhatja a betegség ilyen megnyilvánulásait:
- kifejezett vagy enyhe strabismus,
- a nyelv mozgásának gyenge kontrollja, melynek következtében a betegek ok nélkül kifogásolják a nyelvüket,
- a lenyelés és szívás nehézségei, különösen a fiatalabb gyermekeknél,
- a bőr és a szem pigmentáció megsértése,
- felemelt vagy hajlított,
- giperrefleksiya,
- alvászavarok, különösen a gyermekkorban,
- gyakori saliváció,
- nyomasztó szomjúság,
- túlzottan aktív rágásmozgások,
- hősérzékenység,
- lapos fej,
- fejlett állkapocs,
- sima pálmák.
A betegek nagy részében a vizeletürítés problémái vannak, amelyeket kevéssé ellenőrzik, a finom motoros készségek megsértése, ami nehézséget jelent az önkiszolgáló és a túlsúlyos képzésben. Gyakorlatilag minden betegnél a pubertás később kezdődik, mint egészséges társaiknál.
A angelman-szindrómás gyermekek jónak tartják a szóbeli beszéd megértését és megértését, de nem akarnak részt venni a beszélgetésben, korlátozzák beszédüket a mindennapi életben szükséges több tucat szóra. Ám a felnőttkorban a betegek genetikai patológiás betegeknél fiatalabbak, mint társaik.
Az Angelmann-szindróma tünetei sokoldalúak, ezért a betegség klinikai képe megváltozik az életkorral. A görcsrohamok és az epilepsziás rohamok egyre ritkábbak vagy eltűnnek, a beteg kevésbé izgatottak, alváskészletek.
Komplikációk és következmények
Az Angelman-szindróma egy komoly, gyakorlatilag gyógyíthatatlan kromoszomális patológia, amely megfosztja a betegeket a normális élet lehetőségétől. Mi lesz a baba életét az SA-val, nagymértékben függ a kromoszómális rendellenesség típusától.
A kromoszóma régió többszöröse a legtöbb esetben összeegyeztethetetlen az életével. És még akkor is, ha ezek a betegek nem halnak meg gyermekkorukban, és nem érik el a pubertást, nincs lehetőség gyermekük megtartására.
Az Anghelman-szindrómában leggyakrabban előforduló gének egy részének törlése vagy hiánya akadályozza a gyermeket, hogy megtanuljanak sétálni és beszélgetni. Ilyen gyermekeknél a szellemi retardáció súlyosabb formában jelenik meg, epilepsziás rohamok gyakran előfordulnak, intenzitásuk sokkal erősebb, mint más kromoszómális rendellenességek esetén.
Ha csak egy mutáció van az egyik génen, a megfelelő figyelem és megközelítés alapján a gyerek a tanításban az önkiszolgálás, a kommunikáció és a kommunikáció alapjait taníthatja, bár még mindig elmarad a társaitól.
Az Angelmann-féle szindrómás gyermekek számára, természetüknél fogva jóindulatúak, a fő a szülők szeretete és figyelmessége. Csak ebben az esetben a gyermek képzése gyümölcsöt eredményez, még akkor is kicsi. Természetesen a rendszeres iskolai betegek nem tudnak tanulmányozni az SA-val. Külön osztályokra van szükségük, ahol a gyerekeket először tanítják arra, hogy összpontosítsák figyelmüket, majd fokozatosan megismerik az iskolai ismeretek alapjait.
Diagnostics angelman-szindróma
Az Angelman-szindróma a veleszületett fejlődési patológia. De bizonyos körülmények miatt a gyermekkorban és a kora gyermekkorban diagnosztizálható leggyakrabban nem lehetséges. Ennek oka nem specifikus és enyhe tünetek a csecsemők és a kisgyermekek számára legfeljebb 3 évig. És a betegség előfordulása hazánkban nem olyan nagy, hogy az orvosok megtanulták felismerni azt a hasonlóság között.
Angelman-szindróma csecsemők megnyilvánulhat formájában csökkent izomtónus, ami abban nyilvánul formájában táplálási problémák (gyengeség szopás és nyelési reflex), majd később a tanulási nehézségek gyaloglás (mint a gyerekek sokkal később el járni). Ezek a tünetek a csecsemő fejlődésében mutatkozó eltérések első jelei, amelyek kromoszóma rendellenességgel járhatnak. Erősítse meg ezt a feltevést csak genetikai elemzés.
Különös figyelmet szentelnek azoknak a gyermekeknek, akiknek a szülõi különbözõ genomiális vagy kromoszomális rendellenességeket mutatnak. Miután popervosti betegség nem mutatkozik meg semmilyen módon, és ha a rendellenességek kimutatására időben elkezdett élénken részt vegyenek a gyerek, akkor lehet elérni sokkal nagyobb sikert képzés, megrekedt a betegség progresszióját.
Ha a szülők különböző kromoszomális rendellenességeket mutatnak ki, akkor a genetikai elemzés még a csecsemő születése előtt is megtörténik, mivel a CA az embrionális állapotban kimutatható patológiák egyike.
A genetikai kutatás anyagának gyűjteménye kétféle módon történhet:
- invazív (bizonyos százalékos kockázattal, mert a méhbe való bejutáshoz szükséges a magzatvíz tesztje),
- nem invazív (a csecsemő DNS-elemzése az anya vérével).
Ezután a következő kutatások folynak:
- fluoreszkáló in situ hibridizáció (FISH módszer) - egy speciális festékkel jelölt DNS-próbának a vizsgálandó DNS-hez való kötődését, amit mikroszkópos vizsgálat követ.
- az ube3a gén mutációinak elemzése és a lenyomatoló gének,
- a DNS-metiláció analízise a genetika speciális módszerei segítségével.
A genetikai vizsgálatok pontosan pontos információt adnak a kromoszóma rendellenességek esetén, így a jövőben a szülők előre tudják, hogy mire készülnek. Mindazonáltal vannak kivételek. A betegek egy bizonyos csoportjában, az összes tünetjelző tünet jelenlétében az elemzések eredményei továbbra is normálisak maradnak. Ie rendellenességek kimutatására csak gondosan figyeli a gyermek kora gyermekkorban: hogyan kell enni, amikor elkezdtem járni és beszélni, hogy a láb hajlik meg séta m stb
Amellett, hogy a FISH-módszer közül eljárások diagnosztikai eszköz Angelman-szindróma lehet megkülönböztetni tomográfia (CT vagy MR), hogy segítsen megállapítani a státusz és az agy mérete és elektroencefalogram (EEG), amely megmutatja, hogy az egyes agyterületek működik.
Az orvosok végső diagnózisa általában 3-7 éves korban történik, amikor már a beteg már a tünetek nagy részében és a betegség fejlődésének dinamikája látható.
Milyen tesztekre van szükség?
Megkülönböztető diagnózis
Az Angelman-szindróma genetikai patológia, amely valójában nem rendelkezik specifikus megnyilvánulásokkal. A tünetek nagy része egyaránt jelezheti mind a CA-t, mind az egyéb genetikai patológiákat.
Az Anghelman-szindróma differenciáldiagnózisát a következő patológiákkal végzik:
- Pitt-Hopkins-szindróma (a betegeket a mentális retardáció jellemzi, vidám karakter, mosolyogva, meglehetősen széles és széles szájjal rendelkezik, mikrocefália van). Különbség - a hiperventiláció és a légzés késleltetése az ébrenléti állapotban.
- Kristiansona szindróma (pszichiátriai gyengeségű emberek, akik vidám hajlamban vannak, nem beszélhetnek, mikrocefália, ataxia, görcsök, az izmok önkéntelen mozgása jellemzi őket).
- Mowata-Wilson-szindróma (tünetek: mentális retardáció, epilepsziás rohamok, éles áll, nyitott száj, boldogság arckifejezés, mikrocefália). A különbség nagy távolság a szemek között, a szemek befelé hajolnak, az orr hegye lekerekített, a szárnyak visszafordulnak.
- Kabuki szindróma (jellemző, enyhe vagy közepesen súlyos mentális retardáció, beszédzavar és a motoros készségek, izomgyengeség, görcsök, microcephalia, nagy rések zudami, rendezetlenség). A különbség - a szemöldöke formájában egy íj, egy fordított oldalirányú része az alsó szemhéj, széles szemű, hosszú szem rések hosszú vastag szempillák.
- Rett szindróma (differenciálódás CA-val a nőknél). Tünetek: késleltetett beszédfejlődés, görcsrohamok, mikrocefália. A különbség - nincs boldog kifejezés az arcon, apnea és apraxia támadások vannak, amelyek végül előrehaladnak.
- Szindróma autoszomális recesszív mentális tardatsii 38 (tünetek: mentális retardáció egy észrevehető késedelmet a motorikus képességek és a beszéd, izomgyengeség, táplálási problémák csecsemőkorban, impulzivitás). A különbség az írisz kék színe.
- A MESR 2 gén ismétlődésének szindróma (differenciálódás a SA-val férfiakban). Tünetek: súlyos mentális retardáció, gyermekkori izomgyengeség, beszéd vagy hiány hiánya, epilepszia. Különbségek - progresszív myopathia, folyamatosan visszatérő fertőzések.
- Clifstra-szindróma (tünetek: beszéd- és gondolkodási problémák, izomgyengeség, alvászavarok, figyelemhiány, enyhén nyitott száj, hiperaktivitás, görcsök, ataxia, egyensúlyhiány). Különbségek - lapos arc, rövid orr, széles szemüveg, nagy fordított alsó ajak, agresszió támadások.
- Smith-Magenis szindróma (görcsrohamok, alvászavarok, szellemi és motorfejlesztési zavarok jellemzik). Különbségek - széles és lapos arc, domború homlok.
- Kulena-de Vries-szindróma (enyhe és mérsékelt mentális retardáció, izomgyengeség, görcsös támadások, barátságosság). Különbségek - hosszú arc magas homlokkal, kinyúló fülek, ferde szemek, nagyobb ízületi mobilitás, veleszületett szívbetegségek.
- Philan - McDermid szindróma (tünetek: mentális retardáció, beszédzavar vagy hiány). Különbségek - nagy kezek fejlett izmokkal, izomgyengeség a születés óta, gyenge izzadás.
Angelman szindróma hasonló tünetek „dicsekedni”, és, hogy egy ilyen patológia adenilsuktsinazy hiány, egy autoszomális recesszív szindróma mentális retardáció 1, a kromoszómán 2q23.1 párhuzamos szindróma, haploinszufficiencia gének FOXG1, STXBP1 vagy MEF2C és mások.
Az orvos feladata, hogy pontos diagnózist készítsen, megkülönbözteti az Angelmann-szindrómát a hasonló tünetekkel járó kórtól, és olyan hatékony kezelést ír elő, amely releváns a betegség kialakulásának diagnosztizálására.
Ki kapcsolódni?
Kezelés angelman-szindróma
Az Angelman-szindróma az említett patológiák kategóriájára utal, és arra törekszik, hogy hatékonyan kezelje a gyógyszert a mai napig. A betegség etiológiai kezelése különböző módszerek és eszközök fejlődési szakaszában van, amelyek közül sok még emberben nem tesztelt. Eddig az orvosok, hogy korlátozni kell a tüneti kezelés, hogy segítsen valahogy enyhíteni a sorsát gyermekek és felnőttek báb-szindróma, szenvedő epilepsziás rohamok, nyáladzás, alacsony vérnyomás, alvászavarok.
Ezért csökkenti az epilepsziás rohamok gyakoriságát és erejét egy megfelelően kiválasztott antikonvulzív gyógyszerrel. De a probléma az, hogy a görcsök kezelésére CA különböznek a szokásos epilepsziás rohamok, így jellemzi őket többféle rohamok, és ezáltal enyhíti a feltétel lesz a bevezetése számos gyógyszer.
A legnépszerűbb görcsoldó kezelésére használt Angelman szindróma: valproinsav, topiramát, lamotrigin, levetiracetám, klonazepám és készítményeik. Ritkábban használt gyógyszerek alapján karmazepina, fenitoin, fenobarbitál, etoszuximid, mert néhány közülük válthatna paradox hatás, hogy megerősítse és gyakoriságának növelése epilepsziás rohamok. Ez akkor történik, ha a gyógyszert a monoterápia részeként használják.
A saliváció kezelésére általában két módszert alkalmaznak: gyógyszerek (nyálképződést gátló készítmények) és működőképesek, amelyek a nyálcsatorna újbóli beültetéséből állnak. De a CA esetében ezek a módszerek hatástalanok maradnak, és a kérdés nyitva marad. A szülők és azok, akik ilyen betegeket törődnek, különös figyelmet kell fordítanunk erre a pillanatra, mivel a páciensek általában nem szabályozzák a nyálázást, és néhány egyszerűen nem tud gondoskodni magáról.
Egy másik probléma az alvás rövid időtartama. Gyakran az Angelman-szindrómával szenvedő gyermekek alig több mint 5 órát alszanak, ami negatívan befolyásolja az egész szervezet munkáját. Izgalmas, aktív gyerekek, szerető játékok és kommunikáció (még akkor is, ha igyekeznek magukat nem verbális módon megtenni), észrevehetően fáradtak a naphoz. Ahhoz, hogy jó pihenő legyen, a testnek teljes egészséges alvásra van szüksége, de ez csak a baj vele.
Úgy tűnik, hogy javítsa az alvás az ingerelhető betegeknél kell lennie ahhoz, gyógyszerek nyugtató hatás (fenotiazinek és atípusos antipszichotikumok), megnyugtatja az idegrendszert. De a CA esetében ezeknek a gyógyszereknek a használata negatív hatásokkal teli. Ezért az orvosok inkább mindig fény hipnotikus gyógyszerek, mint például a „melatonin” (természetes hormonális készítmény alapján az alvási hormon), amely így a betegek egy órával lefekvés előtt 1 tabletta, és a „Difenhidramin”. Az adagolás gyakoriságát és dózisát az orvos állapítja meg, a beteg állapotától és életkorától függően.
Néha a angelman-szindrómában szenvedő betegek problémái vannak az emésztés és a széklet esetében. A szék beállításához lazító készítményekkel van lehetőség (jobb, mint egy fitogenezis).
És lehet megközelíteni a problémát másképp, mint ahogy az amerikai orvosok alapján néhány kezelési módszerek autizmus, mert sok a jellemző tünetek az SA, szintén jellemző az autizmus (impulzivitás, akaratlan mozgások, ismétlődő cselekvések, figyelemhiány, problémák a kommunikáció, stb ) .. Azt is megfigyelték, hogy a közigazgatás a hormon szekretin, normalizálja az emésztést, és egy széket, pozitív hatással van a betegek figyelmét, és az oxitocin javítja a gyermekek kognitív képességek és a memória, a helyes viselkedést.
Igaz, néhány hormon elengedhetetlen itt, különösen, ha a gyermekekről van szó. Az Angelman-szindróma viselkedési terápiát, pszichológus-beszédterápiás munkát (a kommunikáció és a jelbeszéd nem verbális módszereinek oktatása) mutat. Az ilyen gyermekek képzéseinek egyéni programon kell alapulnia, különlegesen képzett tanárok, pszichológusok és szülők részvételével. Sajnos ez nem mindenütt lehetséges, és a családok egyedül maradnak a problémájukkal.
Mivel sok kicsi CA-beteg szenved alacsony izomtónusú és ízületi problémákkal, nagy figyelmet fordítanak a fizioterápiás kezelésre. Az orvosok leggyakrabban paraffin alkalmazásokhoz, elektroferózishoz, mágneses terápiához használnak.
Az aktív tonizáló masszázs és a fizioterápia speciális gyakorlata egy idő után segít a beteg gyermeknek lelkére állni és sétálni. Különösen hasznos ebben a tekintetben az aquagymnastics, amelyet CA-ban ajánlott hideg vízben. Növeli az izmok hangját, és tanítja a babát, hogy saját testét, koordinálja a mozgásokat.
Antikonvulzív kezelés
Az Anghelman-szindróma legveszélyesebb tünete az epilepsziás rohamokhoz hasonló roham. Ezt a tünetet a betegek 80% -ánál észlelték, ami azt jelenti, hogy mindegyiküknek hatékony antikonvulzív kezelést kell előírniuk.
A kezelés az epilepsziás rohamok eszközzel végrehajtott, a vitaminok és görcsoldók. Amikor Angelman szindróma, kíséretében görcsös szindróma, hasznos lesz vitaminok B-csoport, valamint a C-vitamin, D és E. De-vitamin terápiát kijelölni saját ebben az esetben nagyon veszélyes, mert a ellenőrizetlen vitaminok csökkenthetik a hatékonyságát antiepileptikumok és provokál új, súlyos és elhúzódó támadásokat.
Az antikonvulzív szerek kiválasztását és a hatékony dózis kijelölését orvosnak kell kezelnie. Azt is eldönti, hogy elegendő lesz-e egy gyógyszer, vagy a betegnek hosszabb ideig kell 2 vagy több gyógyszert szednie .
A legtöbb beteg az orvosok írnak gyógyszereket valproinsav ( „valproinsav”, „Depakinum”, „Konvuleks”, „valparin” et al.), Amely a görcsrohamok megelőzésére, javítja a hangulatot és a mentális állapot a betegek.
A valproinsav tabletta, szirup és injektálható oldatok formájában áll rendelkezésre. A legnépszerűbb gyógyszer a "Depakin" elhúzódó hatású gyógyszer a tablettákban és intravénás beadásra szolgáló megoldás. A gyógyszer adagját az orvos határozza meg egyedileg a beteg súlyától, életkorától és állapotától függően.
Vegyük a gyógyszert étkezés közben napi 2-3 alkalommal. Az átlagos napi adag 20-30 mg a beteg súlya 1 kilogrammjára, a maximális napi 50 mg / kg.
Ellenjavallata. Nem alkalmazzák a máj és a hasnyálmirigy, a hemorrhagiás diatezis, a májgyulladás, a porfiria és a gyógyszer túlérzékenységének megsértésére.
A mellékhatások között megkülönböztethető a kéz remegése, emésztése és széklet, a testtömeg változása.
A "topiramát" a kábítószer-fogyasztók közül választott gyógyszer. Tabletták formájában készül, és a monoterápia részeként és más gyógyszerekkel együtt alkalmazzák.
Alkalmazási mód és dózis. Vigye be a tablettákat belsejében, tekintet nélkül az étkezéshez. A kezdeti napi bevitel felnőtteknek 25-50 mg, a gyermekek esetében 0,5-1 mg / kg. Minden héten az adagot az orvos rendelése szerint növelik.
A gyógyszert nem szabad a terhesség és a laktáció idején szedni, valamint az összetevők iránti fokozott érzékenységet. A gyógyszer számos más mellékhatással jár.
Drugs, hogy az orvos tudja felírni a Angelman szindróma "Klomazepam", "Rivotril" "A lamotrigin", "Seyzar", "Lamictal", "levetiracetám", "Keppra", "Epiterra" et al.
Alternatív kezelés és homeopátia
Az alternatív gyógyszerek, mint a homeopátiás gyógyszerek, természetesen különböznek az összehasonlító biztonságtól, de itt az Angelholm-szindrómával kapcsolatos ilyen kezelés hatékonysága ellentmondásosnak tekinthető.
Bár néhány alternatív kezelés még mindig segít. Az epilepsziás rohamok megállításáról szól. Ebben a tekintetben a gyógynövényes kezelés nagyon hatásos lehet.
Jó hatású a pünkösdi, az édesgyökér és a kacsafű alapú gyógyhatár (a komponenseket egyenlő mennyiségben veszik fel). A füvet lisztre kell őrölni. A recepció kezdetétől számított 2 hét elteltével észlelhető a görcsrohamok gyakoriságának jelentős csökkenése.
Hasznos a görcsökhöz és a levendula főzetéhez (1 teáskanál egy pohár forró vízhez). A készítményt 5 percig forraljuk, és fél órán keresztül ragaszkodunk hozzá. Vegye a gyógyszert egy éjszakán át 14 napig.
Az epilepsziás rohamokra hatásosnak tekintendő víz (vagy alkohol) infúziós anyajegy.
Tól homeopátiás gyógyszerek megelőzésére rohamok Angelman-szindróma lehet használni gyógyszer kifejlesztése kamilla és gyöngyajakot, Acidum hydrocyanicum, Argentum nitricum, Kalium bromatum, Arsenicum album. De szükséges megfontolni, hogy a konkrét esetekben a készítmények hatékony és biztonságos dózisa csak a háziorvosnak nevezhet ki.
Megelőzés
Ahogy az olvasó valószínűleg már megértette, a gének és más kromoszómális rendellenességek megakadályozására a gyógyszer még mindig túl van a hatalommal szemben, valamint a helyzet orvoslására. Ez mindenkinél megtörténhet, hiszen az Angelmann-szindrómás gyermekek egészséges szülőknél is születnek, és a genetika, amely jelenleg egyike a legkevésbé tanulmányozott orvosi ágaknak, még nem tudja megmagyarázni ezt.
Az egyetlen dolog, amit megtehetünk, hogy vállaljuk a terhesség tervezésével kapcsolatos felelősséget, regisztráljuk és megvizsgáljuk időben. De mégis, egy ilyen intézkedés inkább nem lenne profilaktikus, hanem kognitív, mint bármely felmérés. De a fiatal szülők előre tudják, hogy mit készüljenek fel, és pozitív válasz esetén eldöntik, hogy képesek-e ilyen felelősséget vállalni, mint a beteg gyermek felnevelése.
Előrejelzés
Az Anghelman-szindróma prognózisa a kromoszomális rendellenesség és a kimutathatóság időszerűségétől függ. A legnehezebb része azoknak a gyermekeknek, akiknek 15 kromoszóma "hiányzó" gént tartalmaz (törlés). A séta és beszéd valószínűsége ilyen betegekben rendkívül kicsi. A fennmaradó esetek figyelmes megközelítéssel és a gyermeke szeretetével javítható a korrekció.
Az ilyen betegek, sajnos, nem válhatnak a társadalom teljes tagjává, annak ellenére, hogy messze nem hülyék, megértik a beszédet és annak jelentését. Itt csak a kommunikáció problémái vannak az életben. A betegeket a gyerekkori jelnyelvtől lehet tanítani, de nem lehet kényszerű szavakkal kommunikálni. A beszélő betegek lexikonja a mindennapi életben használt szavak minimálisra korlátozott (5-15 szó).
Ami az Anghelman-szindrómában szenvedő betegek várható élettartamát és általános egészségi állapotát illeti, itt a számok ingadoznak. Felnőttkorban a betegek általában olyan egészségügyi problémákkal szembesülnek, mint a scoliosis és az elhízás, amelyek a megfelelő kezeléssel szemben nem életveszélyesek.