A cikk orvosi szakértője

Új kiadványok
Örökletes nephritis (Alport-szindróma) gyermekeknél
Utolsó ellenőrzés: 05.07.2025

Minden iLive-tartalmat orvosi szempontból felülvizsgáltak vagy tényszerűen ellenőriznek, hogy a lehető legtöbb tényszerű pontosságot biztosítsák.
Szigorú beszerzési iránymutatásunk van, és csak a jó hírű média oldalakhoz, az akadémiai kutatóintézetekhez és, ha lehetséges, orvosilag felülvizsgált tanulmányokhoz kapcsolódik. Ne feledje, hogy a zárójelben ([1], [2] stb.) Szereplő számok ezekre a tanulmányokra kattintható linkek.
Ha úgy érzi, hogy a tartalom bármely pontatlan, elavult vagy más módon megkérdőjelezhető, jelölje ki, és nyomja meg a Ctrl + Enter billentyűt.
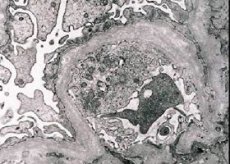
Az örökletes nephritis (Alport-szindróma) egy genetikailag meghatározott örökletes, nem immun glomerulopátia, amely vérvizelésben (néha proteinuriával), a vesefunkció fokozatos romlásában és krónikus veseelégtelenség kialakulásában nyilvánul meg, gyakran szenzorineurális süketséggel és látáskárosodással kombinálva.
A betegséget először 1902-ben írta le L. G. Guthrie, aki egy olyan családot figyelt meg, amelyben több generáción keresztül is megfigyeltek vérvizelést. 1915-ben A. F. Hurst leírta az urémia kialakulását ugyanazon család tagjainál. 1927-ben A. Alport azonosított először halláskárosodást több vérvizeléses rokonnál. Az 1950-es években hasonló betegségben szenvedő szemkárosodásokat írtak le. 1972-ben örökletes vérvizelésben szenvedő betegeknél a vese szövetének morfológiai vizsgálata során Hinglais és munkatársai a glomeruláris bazális membránok egyenetlen tágulását és rétegződését mutatták ki. 1985-ben azonosították az örökletes nephritis genetikai alapját - a IV. típusú kollagén gén mutációját (Fiengold et al., 1985).
A betegség genetikai természetének vizsgálata lehetővé tette számunkra, hogy megállapítsuk, hogy az örökletes nephritis fenotípusos manifesztációinak különbségei (halláskárosodással vagy anélkül) a mutáns gén expressziójának mértékéből adódnak. Így jelenleg minden klinikai változatot egy betegség manifesztációjának tekintünk, és az „örökletes nephritis” kifejezés egyet jelent az „Alport-szindróma” kifejezéssel.
Epidemiológiai vizsgálatok szerint az örökletes nephritis 100 000 gyermekre vetítve 17 esetben fordul elő.
 [
[ Az Alport-szindróma okai
A betegség genetikai alapja a IV. típusú kollagén α-5 láncának génjében bekövetkező mutáció. Ez a típus univerzális a vese, a csiga, a lencsetok, a retina és a szaruhártya bazális membránjaira, amit a kollagénfrakció elleni monoklonális antitestekkel végzett vizsgálatok is bizonyítottak. Nemrégiben felmerült a DNS-próbák alkalmazásának lehetősége az örökletes nephritis prenatális diagnosztikájában.
Hangsúlyozzák a család minden tagjának DNS-próbákkal történő vizsgálatának fontosságát a mutáns gén hordozóinak azonosítása érdekében, ami nagy jelentőséggel bír a betegségben szenvedő családok orvosi és genetikai tanácsadása során. A családok akár 20%-ában sem fordul elő azonban, hogy rokonok szenvednek vesebetegségben, ami a kóros gén spontán mutációinak magas gyakoriságára utal. Az örökletes nephritisben szenvedő betegek többségének családjában vannak vesebetegségben, halláskárosodásban és látászavarban szenvedők; fontosak az egy vagy több őssel rendelkező személyek közötti vérrokon házasságok, mivel rokonok házasságában megnő annak a valószínűsége, hogy mindkét szülőtől ugyanazokat a géneket kapják. Autoszomális domináns, autoszomális recesszív és domináns, X-hez kötött átviteli útvonalakat állapítottak meg.
Gyermekeknél az örökletes nephritis három típusát különböztetik meg leggyakrabban: az Alport-szindrómát, a halláskárosodás nélküli örökletes nephritist és a familiáris jóindulatú hematuriát.
Az Alport-szindróma egy örökletes, halláskárosodással járó nephritis. A vesék, a fül és a szem glomeruláris bazális membránjának kollagénszerkezetének kombinált hibáján alapul. A klasszikus Alport-szindróma génje az X-kromoszóma hosszú karjának 21-22q lókuszában található. A legtöbb esetben domináns módon öröklődik, az X-kromoszómához kötődve. E tekintetben az Alport-szindróma férfiaknál súlyosabb, mivel nőknél a mutáns gén funkcióját a második, sértetlen kromoszóma egészséges allélja kompenzálja.
Az örökletes nephritis kialakulásának genetikai alapja a IV-es típusú kollagén alfa-láncainak génjeiben bekövetkező mutációk. A IV-es típusú G kollagén hat alfa-lánca ismert: az a5- és a6-láncok génjei (Col4A5 és Col4A5) az X-kromoszóma hosszú karján, a 21-22q zónában helyezkednek el; az a3- és a4-láncok génjei (Col4A3 és Col4A4) a 2. kromoszómán; az a1- és a2-láncok génjei (Col4A1 és Col4A2) a 13. kromoszómán.
Az esetek többségében (80-85%) a betegség X-kromoszómához kötött öröklődési mintázatát észlelik, amely a Col4A5 gén deléció, pontmutációk vagy splicing rendellenességek következtében fellépő károsodásával jár. Jelenleg több mint 200 olyan Col4A5 génmutációt találtak, amelyek a IV. típusú kollagén a5-láncainak szintézisének zavaráért felelősek. Ennél az öröklődési típusnál a betegség mindkét nembeli gyermekeknél megnyilvánul, de fiúknál súlyosabb lefolyású.
A IV-es típusú kollagén a3 és a4 láncainak szintéziséért felelős Col4A3 és Col4A4 gének lokuszaiban bekövetkező mutációk autoszomális módon öröklődnek. Kutatások szerint az autoszomális domináns öröklődési típus az örökletes nephritis eseteinek 16%-ában, az autoszomális recesszív típus pedig a betegek 6%-ában figyelhető meg. A Col4A3 és Col4A4 gének mutációinak körülbelül 10 variánsa ismert.
A mutációk eredményeként a IV-es típusú kollagén összeszerelési folyamatai megsérülnek, ami szerkezetének megsértéséhez vezet. A IV-es típusú kollagén a glomeruláris bazális membrán, a cochlea apparátus és a szemlencse egyik fő alkotóeleme, amelynek patológiáját az örökletes nephritis klinikáján lehet kimutatni.
A glomeruláris bazális membrán részét képező IV-es típusú kollagén főként két a1-láncból (IV) és egy a2-láncból (IV) áll, valamint a3, a4, a5-láncokat is tartalmaz. Az X-hez kötött öröklődés során leggyakrabban a Col4A5 gén mutációját az a3-, a4-, a5- és a6-láncok hiánya kíséri a IV-es típusú kollagén szerkezetében, és az o1- és a2-láncok száma megnő a glomeruláris bazális membránban. A jelenség mechanizmusa nem tisztázott, feltételezhető, hogy az ok az mRNS poszttranszkripciós változásai.
Az a3, a4 és a5 láncok hiánya a glomeruláris bazális membránok IV. típusú kollagénjének szerkezetében azok elvékonyodásához és törékenységéhez vezet az Alport-szindróma korai szakaszában, ami klinikailag gyakrabban vérvizelésben (ritkábban proteinuriával vagy csak proteinuriával járó vérvizelésben), halláskárosodásban és lenticonusban nyilvánul meg. A betegség további progressziója a bazális membránok megvastagodásához és permeabilitásának károsodásához vezet a betegség késői szakaszában, az V. és VI. típusú kollagén elszaporodásával, ami a proteinuria növekedésében és a vesefunkció csökkenésében nyilvánul meg.
Az örökletes nephritis alapjául szolgáló mutáció jellege nagymértékben meghatározza annak fenotípusos megnyilvánulását. Az X kromoszóma deléciója és a IV típusú kollagén a5- és a6-láncainak szintéziséért felelős Col4A5 és Col4A6 gének egyidejű mutációja esetén az Alport-szindróma a nyelőcső és a nemi szervek leiomyomatosisával kombinálódik. Kutatási adatok szerint a delécióval járó Col4A5 gén mutációja esetén a kóros folyamat súlyosabb lefolyása figyelhető meg, a vesekárosodás extrarenális manifesztációkkal és a krónikus veseelégtelenség korai kialakulásával, összehasonlítva a gén pontmutációjával.
Morfológiailag az elektronmikroszkópia a glomeruláris bazális membránok (különösen a lamina densa) elvékonyodását és rétegződését, valamint az elektronsűrű granulumok jelenlétét mutatja. A glomeruláris elváltozások heterogének lehetnek ugyanazon betegen belül, a minimális fokális mesangiális elváltozásoktól a glomeruloszklerózisig. Az Alport-szindrómában a glomerulitisz mindig immunnegatív, ami megkülönbözteti a glomerulonephritistől. Jellemző tünetek közé tartozik a tubuláris atrófia kialakulása, a limfohisztiocitás infiltráció, valamint a lipidzárványokkal - lipofágokkal - rendelkező "habsejtek" jelenléte. A betegség előrehaladtával a glomeruláris bazális membránok megvastagodása és kifejezett pusztulása figyelhető meg.
Bizonyos változások figyelhetők meg az immunrendszerben. Az örökletes nephritisben szenvedő betegeknél csökkent az IgA-szint, és hajlamosak az IgM-koncentráció emelkedésére a vérben, az IgG-szint a betegség korai szakaszában emelkedhet, a későbbi szakaszokban pedig csökkenhet. Talán az IgM- és G-koncentráció növekedése egyfajta kompenzációs reakció az IgA-hiányra adott válaszként.
A T-limfocita rendszer funkcionális aktivitása csökken; megfigyelhető az Ig A szintéziséért felelős B-limfociták szelektív csökkenése, az immunitás fagocita kapcsolata megszakad, főként a neutrofilek kemotaxisának és intracelluláris emésztési folyamatainak zavara miatt.
Alport-szindrómás betegek vese biopsziájának vizsgálatakor az elektronmikroszkópos adatok a glomeruláris bazális membrán ultrastrukturális változásait mutatják: elvékonyodás, szerkezeti zavarok és a glomeruláris bazális membránok felhasadása, vastagságának változásával és egyenetlen kontúrokkal. Az örökletes nephritis korai szakaszában a hiba a glomeruláris bazális membránok elvékonyodását és törékenységét határozza meg.
A glomeruláris membránok elvékonyodása kedvezőbb jel, és gyakoribb a lányoknál. Az örökletes nephritis állandóbb elektronmikroszkópos jele az alaphártya megrepedése, amelynek súlyossága korrelál a folyamat súlyosságával.
Az Alport-szindróma tünetei gyermekeknél
Az Alport-szindróma első tünetei izolált húgyúti szindróma formájában leggyakrabban az élet első három évében jelentkeznek. A legtöbb esetben a betegséget véletlenül észlelik. A húgyúti szindrómát a gyermek megelőző vizsgálata során, gyermekfelügyeleti intézménybe való felvétel előtt vagy ARVI során észlelik. ARVI során a vizeletben kimutatható patológia esetén. Az örökletes nephritisben, a szerzett glomerulonephritisszel ellentétben, nincs lappangó időszak.
A betegség kezdeti szakaszában a gyermek egészsége alig romlik, jellemző vonása a húgyúti szindróma tartóssága és ellenállása. Az egyik fő tünet a különböző súlyosságú vérvizelés, amely az esetek 100%-ában megfigyelhető. A vérvizelés mértékének növekedése légúti fertőzések, fizikai aktivitás vagy megelőző oltások után figyelhető meg. A proteinuria a legtöbb esetben nem haladja meg az 1 g/nap értéket, a betegség kezdetén lehet állandó, a folyamat előrehaladtával a proteinuria fokozódik. Időnként a limfociták túlsúlyával járó leukocituria is jelen lehet a vizelet üledékében, ami intersticiális változások kialakulásával jár.
Ezt követően részleges vesefunkció károsodik, a beteg általános állapota romlik: mérgezés, izomgyengeség, artériás hipotenzió, gyakran halláskárosodás (különösen fiúknál), és néha látáskárosodás jelentkezik. A mérgezés sápadtságban, fáradtságban és fejfájásban nyilvánul meg. A betegség kezdeti szakaszában a halláskárosodást a legtöbb esetben csak audiográfiával észlelik. Az Alport-szindrómában a halláskárosodás a gyermekkor különböző időszakaiban jelentkezhet, de leggyakrabban 6-10 éves korban diagnosztizálják. A gyermekek halláskárosodása magas frekvenciákkal kezdődik, jelentős mértékű levegő- és csontvezetést eredményez, a hangvezetési halláskárosodástól a hangérzékelési halláskárosodásig terjed. A halláskárosodás a betegség egyik első tünete lehet, és megelőzheti a húgyúti szindrómát.
Az Alport-szindrómás betegek 20%-ában a látószervek elváltozásai figyelhetők meg. A leggyakrabban észlelt rendellenességek a lencsét érintik: szferofókia, elülső, hátsó vagy kevert lencsekónus, valamint különféle szürkehályogok. Az Alport-szindrómás családokban jelentős a rövidlátás gyakorisága. Számos kutató folyamatosan megfigyel kétoldali perimakuláris elváltozásokat ezekben a családokban élénk fehéres vagy sárgás granulációk formájában a sárgatestben. Ezt a jelet állandó tünetnek tekintik, amelynek magas diagnosztikai értéke van az Alport-szindrómában. KS Chugh és munkatársai (1993) egy szemészeti vizsgálatban az Alport-szindrómás betegeknél az esetek 66,7%-ában a látásélesség csökkenését, 37,8%-ában elülső lencsekónuszt, 22,2%-ában retinafoltokat, 20%-ában szürkehályogot és 6,7%-ában keratokónuszt találtak.
Egyes örökletes nephritisben szenvedő gyermekeknél, különösen veseelégtelenség kialakulásakor, jelentős fizikai fejlődési késés figyelhető meg. A veseelégtelenség előrehaladtával artériás magas vérnyomás alakul ki. Gyermekeknél gyakrabban észlelhető serdülőkorban és idősebb korcsoportokban.
Az örökletes nephritisben szenvedő betegekre a kötőszöveti diszmorfogenezis különféle (több mint 5-7) stigmái jellemzőek. A betegeknél előforduló kötőszöveti stigmák közül a leggyakoribbak a szemek hipertelorizmusa, a magas szájpadlás, a harapási rendellenességek, a fülkagylók rendellenes alakja, a kisujj görbülete a kézen és a "szandálrés" a lábon. Az örökletes nephritisre jellemző a diszmorfogenezis stigmák családon belüli egyenletessége, valamint azok magas gyakorisága a betegség terjedési vonalán áthaladó probandok rokonai között.
A betegség korai szakaszában a részleges vesefunkciók izolált csökkenése észlelhető: aminosavak, elektrolitok szállítása, koncentrációfunkció, acidogenezis, későbbi változások a nefron proximális és disztális részének funkcionális állapotát is befolyásolják, és kombinált részleges zavarok jellemzik őket. A glomeruláris filtráció csökkenése később, gyakrabban serdülőkorban jelentkezik. Az örökletes nephritis előrehaladtával vérszegénység alakul ki.
Így az örökletes nephritist a betegség szakaszos lefolyása jellemzi: először egy látens stádium vagy rejtett klinikai tünetek jelentkeznek, amelyek a húgyúti szindróma minimális változásaiban nyilvánulnak meg, majd a folyamat fokozatos dekompenzációja következik be a vesefunkció csökkenésével, manifeszt klinikai tünetekkel (mérgezés, gyengeség, fejlődési késleltetés, vérszegénység). A klinikai tünetek általában a gyulladásos reakció rétegződésétől függetlenül jelentkeznek.
Az örökletes nephritis különböző korosztályokban jelentkezhet, ami a gén hatásától függ, amely egy bizonyos ideig elnyomott állapotban van.
Osztályozás
Az örökletes nephritisnek három típusa van
- I. lehetőség - klinikailag nephritisként jelentkezik vérvizeléssel, halláskárosodással és szemkárosodással. A nephritis lefolyása progresszív a krónikus veseelégtelenség kialakulásával. Az öröklődés típusa domináns, az X kromoszómához kapcsolódik. Morfológiailag az alaphártya szerkezetének megsértése, elvékonyodása és felhasadása észlelhető.
- II. lehetőség - klinikailag halláskárosodás nélküli vérvizeléssel járó nephritisként jelentkezik. A nephritis lefolyása progresszív, krónikus veseelégtelenség alakul ki. Az öröklődés típusa domináns, az X kromoszómához kapcsolódik. Morfológiailag a glomeruláris kapilláris alaphártya (különösen a laminadensa) elvékonyodása észlelhető.
- III. lehetőség - jóindulatú familiáris hematuria. A lefolyás kedvező, krónikus veseelégtelenség nem alakul ki. Az öröklődés típusa autoszomális domináns vagy autoszomális recesszív. Az autoszomális recesszív öröklődés esetén a betegség súlyosabb lefolyása figyelhető meg nőknél.
Az Alport-szindróma diagnózisa
A következő kritériumokat javasolják:
- legalább két nephropathiás beteg jelenléte családonként;
- a hematuria, mint a nephropathia vezető tünete a probandban;
- halláskárosodás jelenléte legalább egy családtagnál;
- krónikus veseelégtelenség kialakulása egy vagy több rokonnál.
A különféle örökletes és veleszületett betegségek diagnosztizálásában nagy helyet kap a vizsgálat átfogó megközelítése, és mindenekelőtt a gyermek családfájának összeállításakor kapott adatok figyelembevétele. Az Alport-szindróma diagnózisát akkor tekintik érvényesnek, ha a betegnél 4 tipikus tünetből 3-at észlelnek: vérvizelés és krónikus veseelégtelenség jelenléte a családban, neuroszenzoros halláskárosodás jelenléte, látászavar a betegnél, a glomeruláris bazális membrán hasadásának jeleinek észlelése vastagságának változásával és egyenetlen kontúrokkal a biopszia elektronmikroszkópos jellemzői során.
A beteg vizsgálatának tartalmaznia kell klinikai és genetikai kutatási módszereket; a betegség kórtörténetének célzott vizsgálatát; a beteg általános vizsgálatát, figyelembe véve a diagnosztikailag jelentős kritériumokat. A kompenzációs stádiumban a patológia csak olyan szindrómákra összpontosítva mutatható ki, mint az örökletes terhelés, hipotenzió, a diszembriogenezis többszörös stigmái, a húgyúti szindróma változásai. A dekompenzációs stádiumban extrarenális tünetek jelentkezhetnek, mint például súlyos mérgezés, gyengeség, késleltetett fizikai fejlődés, vérszegénység, amelyek a vesefunkció fokozatos csökkenésével jelentkeznek és fokozódnak. A legtöbb betegnél a vesefunkció csökkenésével a következők figyelhetők meg: csökkent acido- és aminogenezis; a betegek 50%-ánál a vesék szekréciós funkciójának jelentős csökkenése; a vizelet optikai sűrűségének korlátozott ingadozása; a filtrációs ritmus zavara, majd a glomeruláris filtráció csökkenése. A krónikus veseelégtelenség stádiumát akkor diagnosztizálják, ha a betegek vérszérumában 3-6 hónapig vagy tovább emelkedett a karbamidszint (több mint 0,35 g/l), és a glomeruláris filtráció a normál érték 25%-ára csökken.
Az örökletes nephritis differenciáldiagnosztikájában elsősorban a szerzett glomerulonephritis hematuriás formáját kell elvégezni. A szerzett glomerulonephritis leggyakrabban akut kezdettel, a fertőzés után 2-3 héttel kezdődik, extrarenális tünetekkel, beleértve a magas vérnyomást az első napoktól (örökletes nephritis esetén ezzel szemben alacsony vérnyomással), a betegség kezdetén csökkent glomeruláris filtrációval, a részleges tubuláris funkciók károsodásával, míg örökletes esetben ezek jelen vannak. A szerzett glomerulonephritis kifejezettebb hematuriával és proteinuriával, fokozott ESR-rel jelentkezik. Diagnosztikai értékkel bírnak a glomeruláris bazális membrán tipikus változásai, amelyek az örökletes nephritisre jellemzőek.
A diszmetabolikus nephropathia differenciáldiagnózisát krónikus veseelégtelenség esetén végzik, a családban klinikailag heterogén vesebetegségeket mutattak ki, és a nephropathia spektruma a pyelonephritistől az urolithiasisig terjedhet. A gyermekek gyakran panaszkodnak hasi fájdalomra és időszakosan vizelés közben, a vizelet üledékében oxalátok jelennek meg.
Örökletes nephritis gyanúja esetén a beteget speciális nefrológiai osztályra kell utalni a diagnózis tisztázása érdekében.
Mit kell vizsgálni?
Hogyan kell megvizsgálni?
Milyen tesztekre van szükség?
Ki kapcsolódni?
Alport-szindróma kezelése
A kezelési rend magában foglalja a nehéz fizikai megterhelés és a friss levegőn való tartózkodás korlátozását. Az étrend teljes értékű, elegendő mennyiségű teljes értékű fehérjét, zsírt és szénhidrátot tartalmaz, figyelembe véve a vesefunkciót. Nagy jelentőséggel bír a krónikus fertőzési gócok felderítése és kezelése. A következő gyógyszereket alkalmazzák: ATP, kokarboxiláz, piridoxin (legfeljebb 50 mg/nap), karnitin-klorid. A kúrákat évente 2-3 alkalommal adják be. Hematuria esetén gyógynövényes gyógyszert írnak fel - csalán, berkenyelé, cickafark.
A külföldi és hazai szakirodalomban beszámolnak a prednizolonnal történő kezelésről és a citosztatikumok használatáról. A hatás megítélése azonban nehéz.
Krónikus veseelégtelenség esetén hemodialízist és veseátültetést alkalmaznak.
Az örökletes nephritis specifikus (hatékony patogenetikai) terápiájára nincsenek módszerek. Minden kezelési intézkedés a vesefunkció csökkenésének megelőzésére és lassítására irányul.
Az étrendnek kiegyensúlyozottnak és magas kalóriatartalmúnak kell lennie, figyelembe véve a vesék funkcionális állapotát. Funkcionális zavarok hiányában a gyermek étrendjének elegendő fehérjét, zsírt és szénhidrátot kell tartalmaznia. Veseműködési zavar jeleinek jelenlétében korlátozni kell a fehérje, szénhidrát, kalcium és foszfor mennyiségét, ami késlelteti a krónikus veseelégtelenség kialakulását.
A fizikai aktivitást korlátozni kell; a gyermekeknek azt tanácsolják, hogy kerüljék a sporttevékenységeket.
Kerülni kell a fertőző betegekkel való érintkezést, csökkenteni kell az akut légúti betegségek kialakulásának kockázatát. Szükséges a krónikus fertőzés gócainak fertőtlenítése. Örökletes nephritisben szenvedő gyermekeknél megelőző oltásokat nem végeznek, az oltás csak epidemiológiai indikációk esetén lehetséges.
Az örökletes nephritis hormonális és immunszuppresszív terápiája hatástalan. Vannak arra utaló jelek, hogy a ciklosporin A és az ACE-gátlók hosszú távú, több éves alkalmazása pozitív hatást fejt ki (a proteinuria csökkenése és a betegség progressziójának lassulása).
A betegek kezelésében olyan gyógyszereket használnak, amelyek javítják az anyagcserét:
- piridoxin - 2-3 mg/kg/nap 3 adagban 4 héten keresztül;
- kokarboxiláz - 50 mg intramuszkulárisan minden második nap, összesen 10-15 injekció;
- ATP - 1 ml intramuszkulárisan minden második nap, 10-15 injekció;
- A-vitamin - 1000 NE/év/nap 1 adagban 2 héten keresztül;
- E-vitamin - 1 mg/kg/nap 1 adagban 2 héten keresztül.
Ez a fajta terápia javítja a betegek általános állapotát, csökkenti a tubuláris diszfunkciókat, és évente háromszor kúrában történik.
A levamisol immunmodulátorként alkalmazható - 2 mg/kg/nap dózisban, heti 2-3 alkalommal, 3-4 napos szünetekkel az adagok között.
Kutatási adatok szerint a hiperbárikus oxigénellátás pozitív hatással van a hematuria és a veseműködési zavar súlyosságára.
Az örökletes nephritis kezelésének leghatékonyabb módszere az időben elvégzett veseátültetés. Ebben az esetben a transzplantáció során nem következik be a betegség kiújulása; az esetek kis százalékában (kb. 5%-ában) nephritis alakulhat ki az átültetett vesében, a glomeruláris bazális membrán antigénjeihez kapcsolódóan.
Ígéretes irány a prenatális diagnosztika és a géntechnológiai terápia. Az állatkísérletek a IV. típusú kollagén alfa-láncok szintéziséért felelős normál gének veseszövetbe történő átvitelének nagy hatékonyságát mutatják, majd a normál kollagénstruktúrák szintézise figyelhető meg.
Előrejelzés
Az örökletes nephritis prognózisa mindig komoly.
Az örökletes nephritis lefolyásának prognosztikailag kedvezőtlen kritériumai a következők:
- férfi nem;
- krónikus veseelégtelenség korai kialakulása a családtagokban;
- proteinuria (napi 1 g-nál több);
- a glomeruláris alaphártyák megvastagodása mikroszkópos vizsgálattal;
- akusztikus neuritisz;
- deléció a Col4A5 génben.
A jóindulatú familiáris hematuria prognózisa kedvezőbb.
Использованная литература