A cikk orvosi szakértője

Új kiadványok
Az alkaptonúria egy veleszületett enzim rendellenesség.
Utolsó ellenőrzés: 04.07.2025

Minden iLive-tartalmat orvosi szempontból felülvizsgáltak vagy tényszerűen ellenőriznek, hogy a lehető legtöbb tényszerű pontosságot biztosítsák.
Szigorú beszerzési iránymutatásunk van, és csak a jó hírű média oldalakhoz, az akadémiai kutatóintézetekhez és, ha lehetséges, orvosilag felülvizsgált tanulmányokhoz kapcsolódik. Ne feledje, hogy a zárójelben ([1], [2] stb.) Szereplő számok ezekre a tanulmányokra kattintható linkek.
Ha úgy érzi, hogy a tartalom bármely pontatlan, elavult vagy más módon megkérdőjelezhető, jelölje ki, és nyomja meg a Ctrl + Enter billentyűt.
Az egyik nagyon ritka anyagcserezavar, az alkaptonuria, a tirozin aminosav anyagcseréjének veleszületett rendellenességeire utal.
Ez a szindróma homogentizat-oxidáz hiánynak, homogentisinuriának, örökletes ochronózisnak vagy fekete vizelet betegségnek is nevezhető.[ 1 ]
Járványtan
A statisztikák szerint az alkaptonuria esetei száma nem haladja meg a kilencet 1 millió emberre vetítve. A legtöbb európai országban pedig 100-250 ezer élveszületésre jut egy eset.
Az európai országok közül kivételt képez Szlovákia (különösen a viszonylag kis északnyugati régió), ahol az alkaptonuria prevalenciája 19 000 újszülöttre vetítve egy eset. Ez valószínűleg annak köszönhető, hogy az ott élő szlovák roma családok körében a beltenyészet (unokatestvérek közötti házasság) szintje a legmagasabb Európában: 10-14%. [ 2 ]
Okoz alkaptonuria
Az alkaptonuria, mint az aromás (homociklusos) α-aminosav tirozin katabolizmusának (metabolikus lebomlásának) veleszületett rendellenességének pontos okai megállapítottak: ez a típusú anyagcserezavar a 3. kromoszómán található több ezer gén egyikének homozigóta vagy összetett heterozigóta mutációinak következménye, pontosabban a kromoszóma hosszú karján található 3q21-q23 lókuszban található HGD géné. Ez a gén a homogentizat-1,2-dioxigenáz májenzim [ 3 ] (más néven homogentizinsav-oxidáz vagy homogentizat-oxidáz) nukleotidszekvenciáit kódolja - egy vastartalmú metalloproteint, amely a szervezetben a tirozin lebontásának egyik szakaszához szükséges. [ 4 ], [ 5 ]
Így az alkaptonuria a homogentizat-1,2-dioxigenáz enzim hibája, pontosabban annak genetikailag meghatározott hiánya vagy teljes hiánya. [ 6 ]
Mivel veleszületett enzimhiányról van szó, az alkaptonuria autoszomális recesszív módon öröklődik, vagyis ahhoz, hogy gyermekeknél alkaptonuria alakuljon ki, mindkét szülőnek rendelkeznie kell az enzim módosított génjével, mivel mindegyikük a rendelkezésre álló két génből csak egy példányt örökít át a gyermekre.
A legfrissebb adatok szerint a HGD gén módosításának több mint kétszáz változata létezik, és leggyakrabban a missense mutációkat, a transzlokációt és a splicingot figyelik meg.
Kockázati tényezők
Az egyetlen kockázati tényező a veleszületett enzimopátia kialakulásában a családi előfordulás és a HGD gén két módosított példányának öröklődése, ha a szülők nem mutatnak alkaptonuriát (a rendellenesség átvitelének kockázata 25%), vagy ha az egyik szülőnél fennáll ez a rendellenesség. [ 7 ]
Pathogenezis
A tirozin kulcsszerepet játszik a fehérjék szintézisében, a kromoproteinek – a melanin bőrpigment –, valamint a pajzsmirigyhormonok és a katekolamin neurotranszmitterek termelésében.
A sejtekben lévő tirozin mennyiségét szabályozó mechanizmus nagyon összetett, és a szervezet a felesleges mennyiségét lebontásával normalizálja. A tirozin katabolizmusának folyamata, mint minden aromás aminosav esetében, többlépcsős és több szakaszban zajlik. A tirozin metabolikus lebontásának minden szakasza egy specifikus enzim részvételével és egy köztes vegyület képződésével történik.
Tehát először az aminosav para-hidroxifenilpiruváttá bomlik le, amely alkaptonná – 2,5-dihidroxifenilecetsavvá vagy homogentizinsavvá – alakul. Ezután az alkaptonnak maleecetsavvá kellene átalakulnia, de ez nem történik meg. [ 8 ]
Az alkaptonuria patogenezise a tirozin-katabolizmus biokémiai reakcióinak megszűnésében áll a homogentizinsav képződésének szakaszában: egyszerűen nincs szükség enzimre a lebontásához – homogentizin-oxidázra.
A homogentizinsavat a szervezet nem hasznosítja, és a vesén keresztül kiválasztódva felhalmozódhat. Ezenkívül benzokinoacetáttá (benzokinonecetsavvá) oxidálódik, amely a szövetek és testnedvek molekuláihoz kötődve melaninhoz hasonló színű biopolimer vegyületeket képez.
Ezen köztes termékek felhalmozódása a szövetben a porcszövet kollagén szerkezetének zavarához vezet, ami csökkenti annak rugalmasságát – az alkaptonuria számos klinikai tünetének megjelenésével és szövődmények kialakulásával.
Tünetek alkaptonuria
Az alkaptonuriát újszülötteknél és csecsemőknél a vizelet sötétedése jellemzi. Levegővel érintkezve a pelenkákon, alsóneműkön és ingeken lévő vizelet sötétbarnára változik; ez a homogentizinsav felhalmozódásának és felszabadulásának köszönhető, amely benzokinoacetáttá oxidálódik. [ 9 ]
Egyéb tünetek hiányában a kisgyermekek alkaptonuriáját gyakran nem ismerik fel időben, mivel a vizelet több órás vizelés után besötétedhet. Egyes adatok szerint a 12 hónapnál fiatalabb, ezzel az enzimhiányossággal született gyermekeknek csak egyötödét azonosítják klinikai környezetben. Ezért nagyon fontos, hogy a szülők odafigyeljenek csecsemőik gondozására.
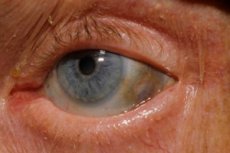
Ezenkívül a korai jelek közé tartozik a szem ínhártyájának, valamint a fülek és az orr porcának pigmentációja (kékes-szürke színe), amit gyakran ochronózisnak neveznek.[ 10 ]
Idővel más tünetek is megjelennek:
- a bőr súlyos pigmentációja az arccsontokon, a hónaljban és a nemi szerveken;
- a ruházat foltosodása, amikor a test izzadt területeivel érintkezik;
- általános gyengeség támadásai;
- rekedt hangon.
Nem szabad elfelejteni, hogy az alkaptonuria és az ochronosis, amint azt fentebb említettük, a tirozin-katabolizmus ugyanazon rendellenességének szinonim nevei.
Juharszirup vizeletbetegség és alkaptonuria. A veleszületett juharszirup vizeletbetegség vagy leucinózis szintén egy anyagcserezavar, ugyanazzal az öröklődési mintázattal rendelkezik, sőt, a mutációk ugyanazon a kromoszómán fordulnak elő, de az elágazó láncú α-keto-sav-dehidrogenáz enzimkomplexet kódoló gént érintik. Emiatt a szervezet nem tudja lebontani a fehérjék bizonyos komponenseit, különösen a leucin, izoleucin és valin aminosavakat. Ennél a betegségnél a vizelet (és a fülzsír) édes illatú; emellett az ilyen típusú szerves acidémia klinikai képéhez tartozik a hipopigmentáció, a vérnyomás ingadozása, görcsrohamok, hányás és hasmenés, a vércukorszint csökkenése, ketoacidózis, hallucinációk stb. A gyermekek halálozási aránya meglehetősen magas; felnőtteknél kezelés nélkül kóma és halál is előfordulhat agyödéma miatt.
Az albinizmust és az alkaptonuriát csak a tirozin „egyesíti”. Az albinizmust, beleértve az oculocutaneous albinizmust is, a melanin pigment termelését befolyásoló genetikai mutációk okozzák. Veleszületett elváltozások figyelhetők meg a 11-es kromoszómán található TYR génben (11q14.3), amely a tirozinázt kódolja, egy réztartalmú melanoszóma enzimet, amely a tirozin anyagcsere-termékein alapuló bőrpigment képződéséhez szükséges. Ez a betegség sokkal gyakoribb, mint az alkaptonuria.
Komplikációk és következmények
A tirozin köztes metabolitjainak – a homogentizin- és a benzokinonecetsavnak – a hatása által okozott alkaptonuria következményei és szövődményei a reaktív pigmentált polimerek lerakódása, a kollagénfibrillumok pusztulása és a porc állapotának romlása (a mechanikai stresszel szembeni ellenállásuk csökkenésével) miatt jelentkeznek.
Az évek során, felnőttkorban, degeneratív ízületi gyulladás és osteoarthritis alakul ki a nagy ízületekben (csípő, keresztcsonti és térdcsonti gerinc); a csigolyaközi terek szűkülnek (különösen az ágyéki és háti gerincben) – meszesedés és oszteofiták képződése közben; a porc alatti csontlemezek szövetsűrűsége csökken, és az alatta lévő csontok kóros átalakulást szenvedhetnek el kinövések és deformációk kialakulásával. [ 11 ]
A szívbillentyűk (aorta és mitrális) és a koszorúerek károsodása is megfigyelhető – a koszorúér-betegség jeleivel, valamint kövek képződésével a vesékben és a prosztatában – ugyanazon meszesedés miatt. [ 12 ], [ 13 ]
Diagnostics alkaptonuria
A veleszületett anyagcserezavarok diagnózisa jellemzően a test biológiai folyadékainak vizsgálatán alapul.
Milyen vizsgálatok és reakciók alapján diagnosztizálható az alkaptonuria? Vizeletvizsgálat szükséges a homogentizinsav kimutatásához és szintjének meghatározásához (normál – napi 20-30 mg, emelkedett – 3-8 g). A vizeletmintát gázkromatográfiával vagy tömegspektrometriával vizsgálják folyadékkromatográfia segítségével; lehetőség van a vas-klorid vizeletben való jelenlétének szűrővizsgálatára. [ 14 ]
Létezik egy gyors diagnosztikai módszer is – az alkapton meghatározása szárított vizeletfoltokban papíron (színintenzitás alapján).
A diagnózis tisztázásakor az instrumentális diagnosztika (radiográfia) magában foglalja az osteoarthritis és más ízületi patológiák radiológiai jeleinek azonosítását a betegeknél.
A diagnózist az örökletes betegségek diagnosztizálására szolgáló molekuláris genetikai módszerekkel, például genetikai vizsgálattal és DNS-szekvenálással igazolják. [ 15 ]
Megkülönböztető diagnózis
A differenciáldiagnózis magában foglalja az újszülött hemokromatózisát és akut májelégtelenségét, melaninuria, akut intermittáló porfíria, hemofagocitás limfohisztiocitózis, primer mitokondriális patológia, reumatoid artritisz, Bechterew-kór.
Ki kapcsolódni?
Kezelés alkaptonuria
Az alkaptonuria fő kezelése az aszkorbinsav nagy dózisainak (legalább napi 1000 mg) orális adagolása. Gyermekeknél ez növeli a homogentizinsav kiválasztását a vizelettel, felnőtteknél pedig csökkenti származékának, a benzokinonecetsavnak a vizeletben való tartalmát, és lassítja kötődését az ízületek kötőszöveti struktúráihoz és a kollagénhez. [ 16 ]
Nyugat-európai klinikákon tesztelik a Nitizinont (Orfalin), egy olyan metabolitcsoportba tartozó gyógyszert, amely gátolja a tirozin-katabolizmus második szakaszát: a para-hidroxifenilpiruvát homogentizinsavvá történő átalakulását. Ennek a farmakológiai szernek az alkalmazása azonban a tirozin felhalmozódásához vezet, és súlyos mellékhatásokat okozhat, beleértve a szaruhártya-homályt és a fényérzékenységet, az orrvérzést és a gyomorvérzést, a májelégtelenséget, a vérkép változásait stb. Mindazonáltal az Egyesült Államokban a Nitizinont az FDA jóváhagyta az I. típusú tirozinémia kezelésére. [17 ], [ 18 ]
Ennek megfelelően az alkaptonuria okozta ízületi problémák esetén fizioterápiás kezelést – edzésterápiát az izomerő növelésére és az ízületi mobilitás javítására, balneoterápiát és peloidterápiát a fájdalom csökkentésére – alkalmaznak.
Bár a tirozint nemcsak táplálékkal biztosítjuk, hanem a szervezet is termeli, az alkaptonuriában szenvedő betegeknek alacsony fehérjetartalmú étrendet kell követniük, és korlátozniuk kell a tirozinban gazdag élelmiszerek, elsősorban a marha- és sertéshús, a tejtermékek (különösen a sajtok), a hüvelyesek, a diófélék és a magvak fogyasztását.
Megelőzés
A génmutációk megelőzése lehetetlen, de a veleszületett rendellenességek magas kockázatával járó gyermekek születésének megelőzése érdekében orvosi genetikai tanácsadásra van szükség a tervezett terhesség előtt azoknak a pároknak, akiknek a családi kórtörténetében örökletes betegségek szerepelnek. [ 19 ]
Előrejelzés
Az alkaptonuria halálos kimenetele nagyon ritka, és a halált súlyos, a szívet és a veséket érintő szövődmények okozhatják. Tehát az alkaptonuriában szenvedők várható élettartama jó.
Az életminőség azonban csökken az ízületekben vagy a gerincben jelentkező intenzív fájdalom miatt, amely a mobilitás jelentős korlátozásával jár, és gyakran progresszív.