A cikk orvosi szakértője

Új kiadványok
Prionok - a prionbetegségek kórokozói
Utolsó ellenőrzés: 06.07.2025

Minden iLive-tartalmat orvosi szempontból felülvizsgáltak vagy tényszerűen ellenőriznek, hogy a lehető legtöbb tényszerű pontosságot biztosítsák.
Szigorú beszerzési iránymutatásunk van, és csak a jó hírű média oldalakhoz, az akadémiai kutatóintézetekhez és, ha lehetséges, orvosilag felülvizsgált tanulmányokhoz kapcsolódik. Ne feledje, hogy a zárójelben ([1], [2] stb.) Szereplő számok ezekre a tanulmányokra kattintható linkek.
Ha úgy érzi, hogy a tartalom bármely pontatlan, elavult vagy más módon megkérdőjelezhető, jelölje ki, és nyomja meg a Ctrl + Enter billentyűt.
A lassú vírusfertőzéseket speciális kritériumok jellemzik:
- szokatlanul hosszú lappangási idő (hónapok, évek);
- a szervek és szövetek, elsősorban a központi idegrendszer specifikus elváltozása;
- a betegség lassú, folyamatos progressziója;
- elkerülhetetlen végzetes kimenetel.

Néhány akut vírusfertőzéseket okozó kórokozó lassú vírusfertőzéseket is okozhat. Például a kanyaróvírus néha SSPE-t okoz, a rubeolavírus pedig progresszív veleszületett rubeolát és rubeola panencephalitist.
Az állatok tipikus lassú vírusfertőzését a visna/madi vírus okozza, amely egy retrovírus. Ez a juhok lassú vírusfertőzésének és progresszív tüdőgyulladásának kórokozója. Az agy fehérállománya elpusztul, bénulás (visna - sorvadás) alakul ki; a tüdő és a lép krónikus gyulladása következik be.
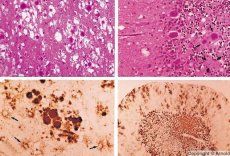
A lassú vírusfertőzésekhez hasonló betegségeket a prionok okozzák - a prionfertőzések kórokozói. A prionbetegségek az emberek és állatok központi idegrendszerének progresszív rendellenességei. Embereknél a központi idegrendszer működése károsodik, személyiségváltozások és mozgászavarok jelentkeznek. A betegség tünetei általában több hónaptól több évig tartanak, és halállal végződnek. Korábban a prionfertőzéseket az úgynevezett lassú vírusfertőzések kórokozóival együtt tekintették.
Néhány prionbetegséget okozó ágens először a nyirokszövetekben halmozódik fel. Az agyba jutó prionok nagy mennyiségben felhalmozódnak, amiloidózist (extracelluláris diszproteinózis, amelyet az amiloid lerakódása jellemez a szövet atrófiájának és szklerózisának kialakulásával) és asztrocitózist (asztrocita neuroglia proliferációja, gliarostok hiperprodukciója) okozva. Fibrillák, fehérje- vagy amiloid-aggregátumok és szivacsos elváltozások alakulnak ki az agyban (fertőző szivacsos agyvelőbántalmak). Ennek eredményeként viselkedésbeli változások, mozgáskoordináció károsodás, halálos kimenetelű kimerültség alakul ki. Nem alakul ki immunitás. A prionbetegségek konformációs betegségek, amelyek a szervezet normális működéséhez szükséges sejtfehérjék helytelen hajtogatása (a helyes konformáció megsértése) következtében alakulnak ki. A prionátvitel útjai változatosak:
- táplálkozási út - fertőzött állati eredetű termékek, nyers szarvasmarha-szervekből származó élelmiszer-adalékanyagok stb.:
- vérátömlesztéssel, állati eredetű gyógyszerek beadásával, szerv- és szövetátültetéssel, fertőzött sebészeti és fogászati eszközök használatával történő átvitel;
- immunbiológiai készítményeken keresztüli átvitel (ismert eset, hogy 1500 juhot fertőzött meg PrP'''-vel beteg juhokból származó agyformol vakcinával).
A kóros prionok, miután bejutottak a bélbe, a vérbe és a nyirokrendszerbe szállítódnak. A lépben, a vakbélben, a mandulákban és más nyirokszövetekben történő perifériás replikáció után a perifériás idegeken keresztül az agyba jutnak (neuroinvazió). A prionok közvetlen behatolása az agyba a vér-agy gáton keresztül is lehetséges. Korábban azt hitték, hogy a központi idegrendszer az egyetlen szövet, amelyben a kóros prionok felhalmozódnak, de megjelentek olyan tanulmányok, amelyek megváltoztatták ezt a hipotézist. Kiderült, hogy a prionok lépben való felhalmozódása összefügg a follikuláris dendritikus sejtek növekedésével és működésével.
 [
[ A prionok tulajdonságai
A prionfehérje normál sejtes izoformáját, amelynek molekulatömege 33-35 kDa, a prionfehérje gén határozza meg (a prion gén - PrNP - a 20. emberi kromoszómán található). A normál gén a sejtfelszínen jelenik meg (a molekula glikoproteinje rögzíti a membránhoz), proteázra érzékeny. Szabályozza az idegimpulzusok továbbítását, a napi ciklusokat, az oxidációs folyamatokat, részt vesz a rézanyagcserében a központi idegrendszerben és a csontvelői őssejtek osztódásának szabályozásában. Ezenkívül a prion gén megtalálható a lépben, a nyirokcsomókban, a bőrben, a gyomor-bél traktusban és a tüszős dendritikus sejtekben.
A kóros prionok elszaporodása
A prionok megváltozott formákká alakulása akkor következik be, amikor a közöttük lévő kinetikailag szabályozott egyensúly felborul. A folyamatot fokozza a kóros (PrP) vagy exogén prion mennyiségének növekedése. A PrP egy normál fehérje, amely a sejtmembránhoz rögzül. A PrP' egy globuláris hidrofób fehérje, amely aggregátumokat képez önmagával és a PrP''-vel a sejtfelszínen: ennek eredményeként a PrP' PrP''-vé alakul, majd a ciklus folytatódik. A PrP'' kóros formája felhalmozódik a neuronokban, szivacsos megjelenést kölcsönözve a sejtnek.
Kuru
Prionbetegség, amely korábban gyakori volt a pápuák (jelentése remegés vagy remegés) körében Új-Guinea szigetének keleti részén. A betegség fertőző tulajdonságait K. Gajdusek bizonyította. A kórokozó rituális kannibalizmus eredményeként terjed étellel - elhunyt rokonok nem kellően átsütött, prionnal fertőzött agyának elfogyasztásával. A központi idegrendszer károsodása következtében a mozgás és a járás károsodik, hidegrázás és eufória ("nevető halál") jelentkezik. A lappangási idő 5-30 évig tart. A beteg egy év után meghal.
Creutzfeldt-Jakob-kór
Prionbetegség, amely demencia, látási és cerebelláris rendellenességek, valamint mozgásszervi rendellenességek formájában jelentkezik, és a Creutzfeldt-Jakob-kór klasszikus változatában 4-5 hónapos betegség után, a Creutzfeldt-Jakob-kór új változatában pedig 3-14 hónap elteltével halálos kimenetelű. A lappangási idő elérheti a 20 évet. A fertőzés különböző módjai és a betegség okai lehetségesek:
- nem kellően hőkezelt állati termékek, például szarvasmarha-szivacsos agyvelőbántalmú tehenek húsának és agyának fogyasztása esetén;
- szövetátültetés során, például szaruhártya-átültetés, vérátömlesztés, hormonok és más állati eredetű biológiailag aktív anyagok használata, catgut használata, szennyezett vagy nem megfelelően sterilizált sebészeti eszközök használata, proszektorális manipulációk;
- a PrR hiperprodukciója és más olyan állapotok esetén, amelyek serkentik a PrR' PrR"-vé alakításának folyamatát
A betegség kialakulhat a prion gén régiójának mutációja vagy inszerciója következtében is. A betegség familiáris jellege gyakori a Creutzfeldt-Jakob-kórra való genetikai hajlam miatt. A Creutzfeldt-Jakob-kór új változatában a rendellenességek fiatalabb korban (átlagéletkor 28 év) alakulnak ki, ellentétben a klasszikus változattal (átlagéletkor 65 év). A Creutzfeldt-Jakob-kór új változatában a kóros prionfehérje nemcsak a központi idegrendszerben, hanem a nyirokretikuláris szövetekben, beleértve a mandulákat is, is felhalmozódik.
Gerstmann-Sträussler-Scheinker szindróma
Örökletes prionbetegség, melyet demencia, hipotónia, nyelési zavar (diszfágia), dysarthria kísér. Gyakran családi eredetű. A lappangási idő 5-30 év. A betegség 50-60 éves korban jelentkezik, időtartama 5-13 év.
Örökletes halálos álmatlanság
Autoimmun betegség, amely progresszív álmatlansággal, szimpatikus hiperaktivitással (magas vérnyomás, hipertermia, hyperhidrosis, tachycardia), tremorral, ataxiával, multiplexszel és hallucinációkkal jár. Az alvás súlyosan megzavarodik. A halál a szív- és érrendszeri elégtelenség progressziójával következik be.
Kaparás
A scrapie (az angol scrape - kaparni szóból) a juhok és kecskék prionbetegsége (rüh), amely a központi idegrendszer károsodásával, progresszív mozgászavarokkal, súlyos bőrviszketéssel (rüh) jelentkezik, és az állat pusztulásával végződik.
Szarvasmarha szivacsos agyvelőgyulladás
Szarvasmarha-betegség, amelyet a központi idegrendszer károsodása, a mozgáskoordináció zavara és az állat elkerülhetetlen pusztulása jellemez. A betegség járványa először Nagy-Britanniában tört ki. Az állatok kóros prionokat tartalmazó hús- és csontliszttel való etetésével függött össze. A lappangási idő 1,5 és 15 év között van. Az állatok agya, gerincvelője és szemgolyói a leginkább fertőzöttek.
Prionbetegségek laboratóriumi diagnosztikája
A diagnosztika során szivacsos elváltozásokat észlelnek az agyban, asztrocitózist (gliózist) és gyulladásos infiltrátumok hiányát. Az agyat amiloidra festik. A prion agyi rendellenességek fehérje markereit az agy-gerincvelői folyadékban mutatják ki (ELISA segítségével). A prion gén genetikai elemzését (PCR) végzik el.
Prionbetegségek megelőzése
A műszerek és a környezeti tárgyak fertőtlenítéséhez autoklávozás (134°C-on 18 percig; 121°C-on 1 órán át), elégetés, további kezelés fehérítővel és egy normál NaCl-oldattal 1 órán át ajánlott. Nem specifikus profilaxis céljából korlátozásokat vezettek be az állati eredetű gyógyszerek használatára, és tilos az állati eredetű agyalapi mirigy hormonok előállítása. A dura mater átültetése korlátozott. A betegek párbeszédfolyadékaival való munka során gumikesztyűt kell használni.