A cikk orvosi szakértője

Új kiadványok
Neuroblasztóma gyermekeknél: okok, diagnózis, kezelés
Utolsó ellenőrzés: 04.07.2025

Minden iLive-tartalmat orvosi szempontból felülvizsgáltak vagy tényszerűen ellenőriznek, hogy a lehető legtöbb tényszerű pontosságot biztosítsák.
Szigorú beszerzési iránymutatásunk van, és csak a jó hírű média oldalakhoz, az akadémiai kutatóintézetekhez és, ha lehetséges, orvosilag felülvizsgált tanulmányokhoz kapcsolódik. Ne feledje, hogy a zárójelben ([1], [2] stb.) Szereplő számok ezekre a tanulmányokra kattintható linkek.
Ha úgy érzi, hogy a tartalom bármely pontatlan, elavult vagy más módon megkérdőjelezhető, jelölje ki, és nyomja meg a Ctrl + Enter billentyűt.
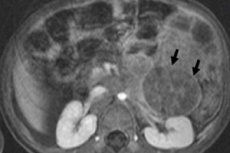
A gyermekgyógyászati onkológiában az egyik leggyakoribb extracranialis daganat a gyermekeknél előforduló neuroblasztóma, amely az idegtaréj neuroblasztjainak, azaz a szimpatikus idegrendszer embrionális (éretlen) idegsejtjeinek embrionális rosszindulatú daganata.
Járványtan
A Nemzetközi Neuroblasztóma Kockázati Csoport (INRG) statisztikái szerint a neuroblasztóma a gyermekek összes onkológiai betegségének körülbelül 8%-át teszi ki világszerte, és a harmadik helyen áll az előfordulásban a leukémia és az agydaganatok után.
Más adatok szerint a neuroblasztóma a csecsemőknél előforduló összes rákos megbetegedés körülbelül 28%-át teszi ki. A neuroblasztóma eseteinek több mint egyharmadát egy év alatti gyermekeknél diagnosztizálják; a diagnózis átlagos életkora 19-22 hónap. A diagnosztizált esetek több mint 90%-a két-öt éves gyermekeknél fordul elő (a fiúk túlnyomó többségében); a csúcs előfordulási gyakoriság két-három éves korban figyelhető meg, az öt év feletti gyermekeknél az esetek kevesebb mint 10%-át teszik ki.
Okoz neuroblasztómák
A neuroblasztóma okainak tanulmányozása során a kutatók arra a következtetésre jutottak, hogy ez a daganat gyermekeknél az embriogenezis vagy a korai posztnatális fejlődés során bekövetkező szórványos genetikai mutációk miatt alakul ki. De hogy mi okozza ezeket a génváltozásokat, az ismeretlen, mivel teratogén környezeti tényezők hatását nem azonosították.
Ezek a daganatok bárhol előfordulhatnak, beleértve a mediastinumot, a nyakat, a hasat, a mellékveséket, a veséket, a gerincet és a medencét.
Ritka esetekben a csecsemőknél előforduló neuroblasztóma örökletes mutációval társulhat. Különösen a 2-es kromoszómán található CD246 membránfehérje génjének – az ALK tirozin-kináz enzimnek – a mutációja, amely biztosítja a sejtek közötti kommunikációt és fontos szerepet játszik az idegrendszer működésében; illetve a PHOX2B fehérje génjének (a 4-es kromoszómán) mutációja, amely részt vesz az idegsejtek érésében.
A neuroblasztóma összefüggésben állhat az 1-es típusú gyermekkori neurofibromatózissal,a Beckwith-Wiedemann szindrómával és a hiperinzulinémiás hipoglikémiával (nesidioblastosis pancreatitis) is.
Kockázati tényezők
Manapság az öröklődést a neuroblasztóma kialakulásának kockázati tényezőjeként ismerik el gyermekeknél - a daganat jelenléte a családi kórtörténetben, valamint a méhen belüli fejlődés során bekövetkező génmutációkkal összefüggő veleszületett rendellenességek. Ez különösen igaz a különböző szervekben több daganat kialakulásának eseteire.
A kutatók nem azonosítottak olyan exogén tényezőket, amelyek növelik a daganat kockázatát.
Pathogenezis
A neuroblasztómák kialakulásának mechanizmusát a velőlemezsejtek differenciálódásának és érésének zavarai okozzák – ezek a kétoldali sejtvonalak az emberi embrió ektodermális csíralemezéből a velőcső szélein alakulnak ki. Ezek a sejtek vándorolnak (mozognak) és sokféle sejtté differenciálódnak: érzékszervi és autonóm neuronokká, neuroendokrin sejtekké és a mellékvesevelő sejtjeivé, az arcporc és a csontok sejtjeivé, valamint pigmentsejtekké.
A neuroblasztómában a migrált neuroblasztok nem érnek, hanem tovább nőnek és osztódnak, tumort képezve. Kialakulásának patogenezise a következő génmutációkkal jár:
- a kromoszóma-szekvenciák egy részének duplikációjával vagy az LMO1 gén szegmenseinek duplikációjával a 11. kromoszómán, amely az RBTN1 fehérjét kódolja az embrió neurális taréjsejtjeiben;
- az 1q21.1 kromoszómán található NBPF10 gén kópiaszámának megváltozásával, amely a DUF1220 fehérjét kódolja, amely az emberi idegi őssejtek proliferációját szabályozza. Ezek a rendellenességek vagy a kromoszóma megduplikálódásához, vagy deléciójához – a DNS egy részének hiányához – vezetnek;
- az ATRX tumorszupresszor gén változásaival (az Xq21.1 kromoszómán);
- a 2. kromoszómán található N-Myc transzkripciós faktor gén további másolatainak (amplifikációjának) jelenlétével, amely az egyik transzkripciós faktort (DNS-kötő fehérje) kódolja, amely más gének aktivitását szabályozza, és a prekurzor sejtek proliferációját szabályozza a magzat szöveteinek és szerveinek képződéséhez szükséges fehérjék képződése során. Ennek a génnek az amplifikációja onkogénné alakítja, ami a sejtciklus zavarát, a sejtek fokozott proliferációját és a tumorképződést idézi elő.
Tünetek neuroblasztómák
A neuroblasztóma első jelei nem specifikusak, és magukban foglalhatják az étvágytalanságot (és a fogyást), az evés közbeni fáradtságot, a lázat és az ízületi fájdalmat.
A klinikai tünetek az elsődleges daganat helyétől és az áttétek jelenlététől függenek (amelyek az esetek 60-73%-ában fordulnak elő).
Nagyon gyakran az elsődleges neuroblasztóma a mellékvesevelőben lokalizálódik, amelynek eredete hasonló az idegsejtekéhez. Egy év alatti gyermekeknél a mellékvese neuroblasztómát az esetek 35-40%-ában diagnosztizálják. Tünetei közé tartozik a hasi fájdalom, láz, fogyás, csontfájdalom, vérszegénység vagy egyidejűleg Pepper-szindróma: diffúz májkárosodás súlyos hepatomegaliával és légzési distressz szindrómával.
A retroperitoneális neuroblasztóma vagy retroperitoneális neuroblasztóma gyermekeknél, ahogy növekszik, nyomást gyakorol a hólyagra vagy a belekre, ami vizelési vagy székletürítési problémákat, a lábak duzzanatát okozhatja (fiúknál a herezacskó megduzzad).
Gyermekeknél a mediastinum neuroblasztómája (mediastinális neuroblasztóma) gyakran nyomja a vena cava superiort, és ez az arc, a nyak, a karok és a mellkas felső részének duzzanatát okozhatja (a bőr kékes-vöröses lesz, bőr alatti csomókkal). Köhögés és zihálás, légzési problémák (légszomj) vagy nyelési problémák (diszfágia) jelentkeznek; megnagyobbodott nyirokcsomók figyelhetők meg a nyakon, a kulcscsont felett és a hónaljban.
A tumorsejtek csontvelőbe történő terjedése vérszegénységhez, trombocitopéniához és leukopéniához vezet, vérzési hajlammal.
A periorbitális területen áttétek esetén sötét karikák vagy zúzódások jelennek meg a szem körül. Az ilyen daganat fejfájást és szédülést, exophthalmiát (a szemgolyók kidudorodását), valamint az idegvégződések összenyomódása miatt - a szemhéjak lelógását (ptosis) és a pupillák méretének csökkenését (miosis) is okozhatja.
A hasi neuroblasztóma vagy a hasüreg neuroblasztómája gyermekeknél tapintható tömítések kialakulásához vezet a hasban, annak puffadásához, étvágytalansághoz, székrekedéshez és megnövekedett vérnyomáshoz. A gerincvelőt vagy az ideggyökeret nyomó daganat a végtagok zsibbadását és gyengeségét, az állás, a kúszás vagy a járás képtelenségét okozhatja. Ha a csontok érintettek, csontfájdalom jelentkezhet.
A hasüregben lévő 3-4. stádiumú, nyirokcsomó-károsodással járó daganat esetén a daganatsejtek bejuthatnak a vese parenchymába, majd gyermekeknél kiterjedt vese neuroblasztóma alakul ki, ami a funkcióinak megzavarásához vezet.
Szakaszai
- Az 1. stádiumú neuroblasztóma egy elsődleges daganat, amely a test egyetlen területére lokalizálódik és izolálódik; a két oldali nyirokcsomók nem érintettek.
- Neuroblasztóma 2. stádium. A 2A stádiumban az elsődleges daganat egyetlen területre korlátozódik, de nagy; kétoldali nyirokcsomók nem érintettek. A 2B stádiumban a daganat oldalán található nyirokcsomók áttétpozitívak.
- Neuroblasztóma 3. stádium: az elsődleges daganat áthatol a gerincvelőn vagy a test középvonalán, egyoldali vagy kétoldali áttétek találhatók a nyirokcsomókban.
- Neuroblasztóma 4. stádium: a daganat átterjedt a távoli nyirokcsomókra, csontvelőre, csontokra, májra vagy más szervekre. A 4S stádiumot egy év alatti gyermekeknél határozzák meg, akiknél lokalizált primer daganat alakul ki, és a bőrbe, májba vagy csontvelőbe terjed.
Nemzetközi Neuroblasztóma Kockázati Stádiumbeosztási Rendszer (INRGSS)
Az INRGSS képalkotó vizsgálatokon alapuló kockázati tényezőket (IDRF) használ, amelyek képalkotó vizsgálatokon látható tényezők, és azt jelenthetik, hogy a daganatot nehezebb lesz eltávolítani.
Az INRGSS a neuroblasztómákat 4 szakaszra osztja:
- L1: A daganat nem terjedt át onnan, ahol kiindult, és nem nőtt létfontosságú struktúrákká. A test egyetlen részére korlátozódik, például a nyakra, a mellkasra vagy a hasra.
- L2: A daganat nem terjedt át (nem adott áttétet) messzire a kiindulási helyétől (például a has bal oldaláról a mellkas bal oldalára növekedhetett), de legalább egy IDRF-e van.
- M: A daganat áttétet adott a test egy távoli részébe (kivéve az MS stádiumú daganatokat).
- MS: 18 hónaposnál fiatalabb gyermekek áttétes betegsége, amelyben a rák csak a bőrre, a májra és/vagy a csontvelőre terjedt ki.
Komplikációk és következmények
A neuroblasztómát a következő szövődmények és következmények jellemzik:
- terjedés (metasztázis) a nyirokcsomókba, a csontvelőbe, a májba, a bőrbe és a csontokba;
- gerincvelő-kompresszió (ami fájdalmat okozhat és bénuláshoz vezethet);
- paraneoplasztikus szindróma kialakulása (a tumor által kiválasztott bizonyos vegyi anyagok, valamint a sejtjei által expresszált GD2 antigén disialogangliosid hatása miatt), amely gyors, akaratlan szemmozgásokban, koordinációs zavarokban, izomgörcsökben és hasmenésben nyilvánul meg;
- a primer terápia befejezése utáni relapszusok (ahogyan azt a klinikai gyakorlat mutatja, a nagy kockázatú neuroblasztómák az esetek 50%-ában relapszussal rendelkeznek).
Diagnostics neuroblasztómák
A gyermek neuroblasztóma gyanújának diagnózisa vizsgálatot, laboratóriumi vizsgálatokat és képalkotást igényel.
Vér- és vizeletvizsgálatot végeznek katekolaminok (norepinefrin és dopamin) és homovanillin- vagy vanilil-mandulasavak (e hormonok anyagcseréje során keletkeznek) kimutatására; vérvizsgálatot neurospecifikus enolázra, enzimhez kötött immunszorbens vizsgálatot (ELISA) végeznek a vérszérumból, és csontvelő-elemzést végeznek (amelyből mintát vesznek aspirációs punkcióval). DNS-tesztet végeznek a mutációk kimutatására, és biopsziát a daganatszövet citomorfológiai vizsgálatára.
A biopsziás minták levétele után azokat laboratóriumba küldik, ahol egy patológus (egy olyan orvos, aki speciális képzésben részesült a rákos sejtek azonosításában) mikroszkóp alatt vizsgálja meg. Gyakran speciális laboratóriumi vizsgálatokat is végeznek a mintákon annak megállapítására, hogy a daganat neuroblasztóma-e.
Ha neuroblasztómáról van szó, a laboratóriumi vizsgálatok segíthetnek meghatározni, hogy milyen gyorsan növekedhet vagy terjedhet a daganat, valamint hogy milyen kezelések lehetnek a leghatékonyabbak.
A műszeres diagnosztika ultrahanggal, röntgennel, MRI-vel vagy CT-vel, 18F-fluorodeoxiglükóz bevezetésével végzett PET-tel vagy metajódbenzilguanidinnal végzett MIBG-szcintigráfiával vizualizálja a daganatot. [ 1 ]
Megkülönböztető diagnózis
A differenciáldiagnózis magában foglalja a jóindulatú ganglioneurómát, a ganglioneuroblasztómát, a rabdomioszarkómát és a nefroblasztómát.
Ki kapcsolódni?
Kezelés neuroblasztómák
Neuroblasztóma esetén a kezelés a beteg kockázati csoportjától (a tumorfolyamat stádiumától), a tumor lokalizációjától, a tumorsejtek genomiális jellemzőitől és a gyermek életkorától függ. Tartalmazhat monitorozást, műtétet, kemoterápiát, sugárterápiát, immunterápiát és vérképző őssejt-transzplantációt.
A gyermekek neuroblasztómájának neoadjuváns vagy adjuváns (pre- vagy posztoperatív) kemoterápiáját, mint bármely rákellenes kemoterápiát, kúrákban adják: a gyógyszert több napon keresztül adják be, majd szünetet tartanak, hogy a szervezet regenerálódhasson. A ciklusokat általában három-négy hetente ismétlik.
A következő gyógyszereket (és azok kombinációit) alkalmazzák: ciklofoszfamid, ciszplatin vagy karboplatin, doxorubicin (adriamicin), vinkrisztin, etopozid.
A kemoterápiás gyógyszerek gyakori mellékhatásai közé tartozik a hajhullás, étvágytalanság, fáradtság, hányinger és hányás, szájfekélyek, hasmenés vagy székrekedés. A kemoterápia károsíthatja a csontvelőt és csökkentheti a vérsejtek számát.
A célzott immunterápia (a GD2 tumor antigén ellen irányul) monoklonális antitestek (anti-GD2 MAb) csoportjába tartozó gyógyszereket, a Dinutuximabot (Unituxin) és a Naxitamabot használja. Ezeket intravénásan, hosszan tartó infúzióban adják be, granulocita-makrofág kolónia stimuláló faktorral (citokin GM-CSF) és interleukin-2-vel kombinálva.
Ezen gyógyszerek mellékhatásai közé tartozik a fájdalom (gyakran nagyon súlyos), csökkent vérnyomás, megnövekedett pulzusszám, légszomj (a légutak esetleges duzzanatával), megnövekedett láz, hányinger, hányás és hasmenés, valamint a vér sejtes és ásványi összetételének változásai.
A nagy dózisú kemoterápia és őssejt-transzplantáció utáni rákkiújulás kockázatának csökkentése érdekében a magas kockázatú neuroblasztómában szenvedő gyermekeket szisztémás retinoidokkal, 13-cisz-retinoinsavval (izotretinoin) kezelik. [ 2 ]
A neuroblasztóma sebészeti kezelése – daganat eltávolítása, például nyílt adrenalectomia vagy laparoszkópos mellékvese neuroblasztóma reszekció; limfektómia (az érintett nyirokcsomók eltávolítása) stb. [ 3 ]
Magas kockázatú neuroblasztóma esetén sugárterápia alkalmazható.[ 4 ]
Megelőzés
Tekintettel a neuroblasztóma okaira gyermekeknél, az egyetlen megelőző intézkedés lehet a genetikai tanácsadás a terhesség tervezésekor. De szem előtt kell tartani, hogy ez a daganat csak az esetek 1-2%-ában kapcsolódik örökletes mutációkhoz.
Előrejelzés
Az infantilis neuroblasztóma képes spontán visszafejlődni.
Prognosztikai markerek
- A magas kockázatú daganatok, valamint a neuroblasztóma minden korcsoportban és minden stádiumban (a 4S stádium kivételével) szenvedő gyermekeknél – az N-MYC gén fokozott expressziójával és az N-Myc onkogén amplifikációjával – kedvezőtlen prognózissal rendelkeznek, ami befolyásolja a várható élettartamot.
- Azok a tumorsejtek, amelyekből az 1-es vagy 11-es kromoszóma bizonyos részei hiányoznak (1p vagy 11q delécióként ismertek), rosszabb prognózissal járnak. A 17-es kromoszóma egy plusz részének megléte (17q nyereség) szintén rosszabb prognózissal jár.
- A nagy mennyiségű DNS-t tartalmazó neuroblasztóma sejtek jobb prognózissal rendelkeznek, különösen a 2 év alatti gyermekek esetében.
- A több neurotrofinreceptorral, különösen a TrkA idegnövekedési faktor receptorral rendelkező neuroblasztómák jobb prognózissal rendelkeznek.
Túlélés gyermekkori onkológiai csoport (COG) kockázati csoportja szerint
- Alacsony kockázatú csoport: Az alacsony kockázatú csoportba tartozó gyermekek 5 éves túlélési aránya meghaladja a 95%-ot.
- Közepes kockázati csoport: A közepes kockázati csoportba tartozó gyermekek 5 éves túlélési aránya 90% és 95% között van.
- Magas kockázatú csoport: A magas kockázatú csoportba tartozó gyermekek 5 éves túlélési aránya körülbelül 50%.
A gyermekkori rákos halálesetek körülbelül 15%-át neuroblasztóma okozza. Ennél a magas kockázatú rosszindulatú daganatnál a hosszú távú túlélés esélye nem haladja meg a 40%-ot. Az ötéves túlélési arány 67-74%, az egy-négy éves korcsoportban 43%, az élet első évében diagnosztizált neuroblasztóma esetében pedig több mint 80%.
Использованная литература